




El concepto de «enfermedad autoinflamatoria» aparece por vez primera en la literatura médica en 1999, propuesto por el Dr. Daniel L. Kastner, del National Institute of Arthritis, Muskuloskeletal and Skin Diseases (NIAMS), para definir un nuevo mecanismo etiopatogénico común a una serie de enfermedades caracterizadas por la aparición de episodios febriles e inflamatorios recurrentes1. Ya desde su definición el concepto de autoinflamación se contrapuso al de autoinmunidad, de tal manera que las enfermedades autoinflamatorias se caracterizan típicamente por la ausencia de autoanticuerpos a títulos elevados y/o células T específicas de antígeno. En la actualidad, las enfermedades autoinflamatorias están consideradas paradigma de enfermedad humana debida a una desregulación de la respuesta inmunitaria innata, y los principales mecanismos efectores son los neutrófilos y los monocitos, así como el equilibrio entre citocinas proinflamatorias y antiinflamatorias2.
Dentro del concepto general de enfermedades autoinflamatorias, se puede distinguir una serie de entidades clínicas independientes, con un claro patrón de herencia mendeliano, que en la actualidad se agrupan bajo el epígrafe de «enfermedades autoinflamatorias heredita-rias»2. Los trabajos de diferentes investigadores durante los últimos años han permitido identificar las bases genéticas de cada una de estas enfermedades, lo que ha posibilitado, por una parte, la aplicación de análisis genéticos en la practica clínica diaria para alcanzar un diagnóstico definitivo y, por otra, el establecimiento de abordajes terapéuticos individualizados para cada una de estas entidades.
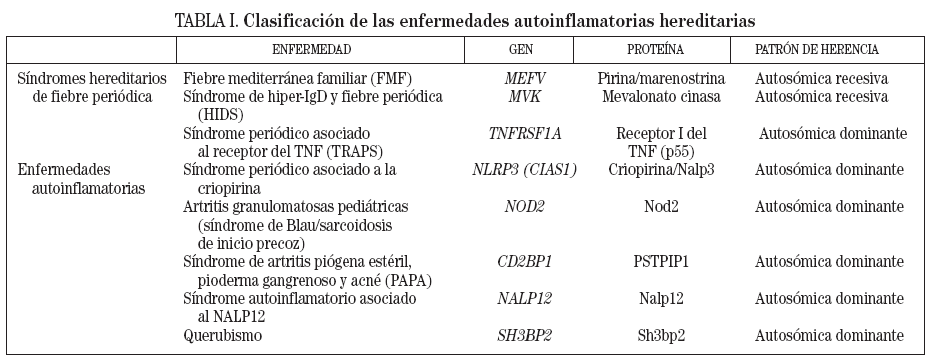
En función de su baja prevalencia, todas las enfermedades autoinflamatorias hereditarias deben ser consideradas enfermedades raras. No obstante, no todas son igual de «raras», y la fiebre mediterránea familiar es la más frecuente de todas ellas, tanto mundialmente como en nuestro país3. En la tabla I se presenta una clasificación de dichas enfermedades, junto con sus patrones de herencia y los genes que causan cada una de ellas.
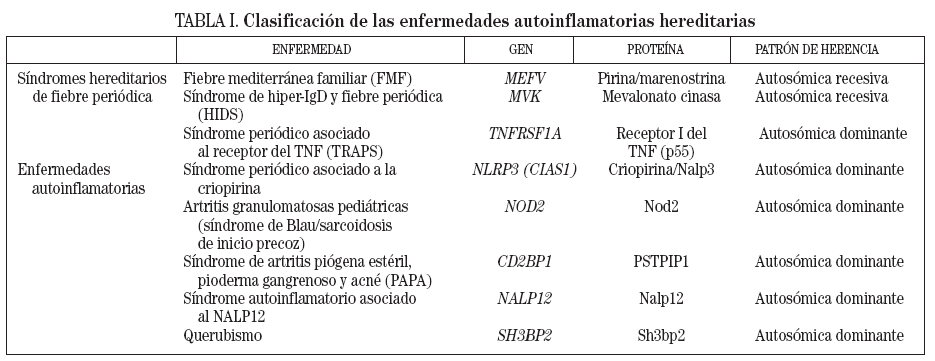
TABLA I. Clasificación de las enfermedades autoinflamatorias hereditarias
Dado que el presente trabajo tiene por objeto la descripción de las enfermedades autoinflamatorias con una importante componente de afección cutánea, a continuación se presentarán en detalle sólo algunas de las enfermedades incluidas en la tabla I; remitimos al lector interesado en todas ellas a la lectura de las diferentes revisiones que se incluyen en el apartado de bibliografía.
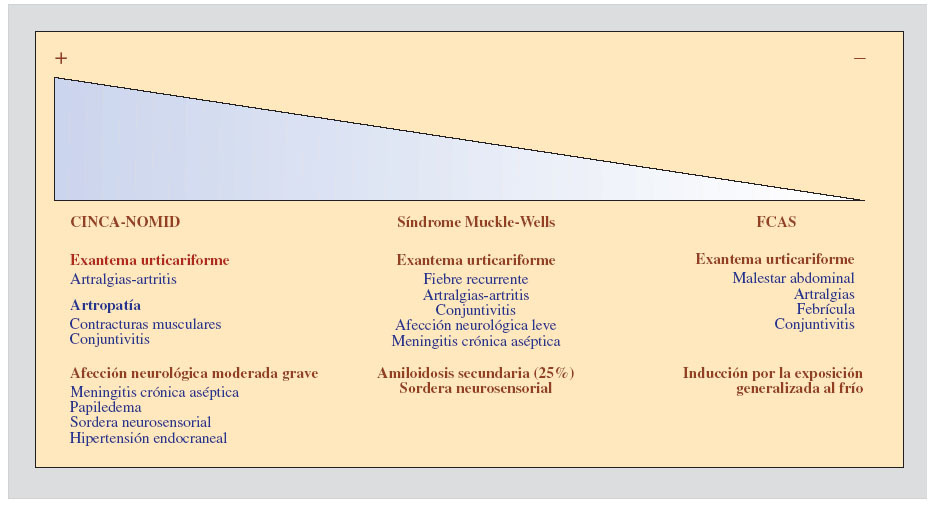
SÍNDROMES PERIÓDICOS ASOCIADOS A CRIOPIRINAEl acrónimo CAPS (del inglés cryopyrin-associated periodic syndromes) fue acuñado para definir una serie de entidades clínicas, aparentemente diferentes, que tienen como mecanismo etiopatogénico la presencia de mutaciones en el gen NLRP3 (también denominado CIAS1, PYPAF1 o NALP3) que codifica la proteína criopirina4. Hoy en día se considera que cada una de estas entidades clínicas representa un grado de severidad dentro de un espectro continuo, de los que el síndrome autoinflamatorio familiar inducido por el frío (familial cold-induced autoinflammatory syndrome [FCAS]), también conocido como urticaria familiar inducida por el frío, la forma más leve, el síndrome de Muckle-Wells la forma intermedia, y el síndrome CINCA (chronic infantile neurologic, cutaneous and articular), también denominado NOMID (neonatal-onset multisystem inflammatory disease), la forma más grave3. Desde un punto de vista clínico, todas estas entidades tienen las siguientes características comunes y una serie de características diferenciales de cada síndrome, que se detallan en la figura 1.
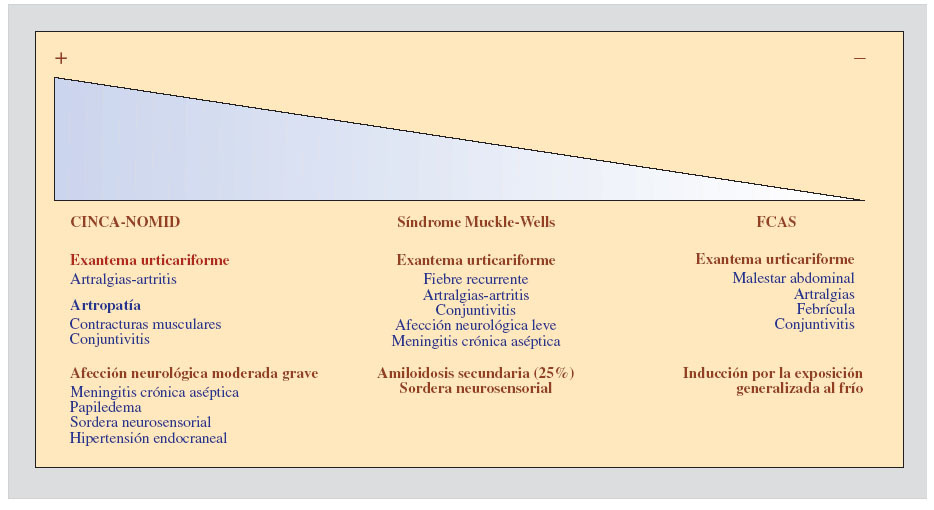
Figura 1. Espectro de severidad clínica creciente de los síndromes periódicos asociados a criopirina. Se resaltan en negrita las características principales de cada entidad.
Todas presentan mutaciones en el gen NLRP3, con un patrón de herencia autosómico dominante3. En este aspecto, debemos señalar la elevada incidencia de casos esporádicos, sin historia familiar de la enfermedad, que se deben a mutaciones de novo, que aparecen por primera vez en el enfermo. Todas se presentan a una edad de inicio muy temprana, habitualmente en los primeros días o semanas de vida.
La aparición de un exantema urticariforme, no pruriginoso, que afecta a áreas importantes de la superficie corporal habitualmente es la primera manifestación clínica en aparecer. Este tipo de manifestación cutánea y el carácter hereditario de la enfermedad han sido las razones por las cuales dichos síndromes han recibido en el pasado la denominación de síndromes urticariformes familiares.
Durante la fase de actividad de la enfermedad, en el hemograma se detecta una leucocitosis a expensas de neutrofilia, trombocitosis e importante reacción de fase aguda, caracterizada por incrementos notables de la VSG y de los valores plasmáticos de proteína C reactiva, fibrinógeno, haptoglobina, ferritina y proteína sérica del amiloide.
Todas las formas clínicas presentan excelentes respuestas clínicas y bioquímicas a los fármacos bloquea-dores de la interleucina (IL) 1β3.
EtiopatogeniaLa base genética del síndrome de Muckle-Wells y el síndrome FCAS se identificó en 2001, cuando el equipo del Dr. Hal M. Hoffman descubrió un nuevo gen (denominado entonces CIAS1 y conocido en la actualidad como NLRP3) y demostró la existencia de mutaciones en dicho gen que segregan con la enfermedad en familias afectas de FCAS y de síndrome de Muckle-Wells5. Un año más tarde, un equipo de investigadores franceses identificó mutaciones en el mismo gen en pacientes afectos del síndrome CINCA-NOMID, y en ese momento se estableció el concepto de CAPS y la existencia de un espectro de gravedad6.
Desde entonces se han identificado más de 60 mutaciones asociadas a enfermedad, localizadas en su práctica totalidad en el exón 3 de dicho gen, disponibles en la base de datos on-line INFEVERS (http://fmf.igh.cnrs.fr/ infevers). A pesar de los esfuerzos realizados, no se ha constatado una estrecha relación genotipo-fenotipo, y se ha evidenciado exclusivamente que una misma mutación no origina fenotipos clínicos extremos7,8.
El gen NLRP3 se expresa en neutrófilos, monocitos y condrocitos y codifica una proteína denominada criopirina (o Nalp3). Dicha proteína pertenece a la familia de receptores citoplásmicos NACHT-LRR (NLR), importantes en la respuesta inmunitaria innata e inflamatoria, en cuanto desempeñan un papel importante en el reconocimiento intracelular de patógenos y señales de alerta9,10. Asimismo, la criopirina forma parte constitutiva del inflamasoma, que es una estructura citoplásmica constituida por múltiples proteínas diferentes (NALP, ASC, caspasas proinflamatorias, CARDINAL) que se asocian o disocian en función de la presencia o ausencia de determinados estímulos. Una vez ensamblado, el inflamasoma tiene por objeto generar la forma activa de caspasa1, la cual a su vez interviene en el procesamiento y la generación de las formas activas de las IL proinflamatorias IL-1β, IL-18 e IL-3311,12.
En la actualidad se ha propuesto que las mutaciones del gen NLRP3 asociadas a los diferentes síndromes CAPS son mutaciones que generan una proteína hiperfuncionante, cuya consecuencia última es la producción incrementada de las citocinas proinflamatorias anteriormente comentadas10.
TratamientoLos abordajes terapéuticos empleados en el pasado han sido habitualmente diferentes en función de la gravedad del cuadro clínico. Así, mientras en las formas más leves (FCAS) las medidas han sido conservadoras, evitando en lo posible los cambios bruscos de temperatura y administrando antihistamínicos o antiinflamatorios no esteroideos, en las formas graves ha sido preciso el empleo de corticoides a dosis altas, con buenas respuestas clínicas, pero con los consabidos efectos secundarios de tratamientos prolongados, especialmente en pacientes pediátricos.
Desde el descubrimiento del papel de la IL-1βen la fisiopatología de dichos síndromes CAPS, los agentes biológicos bloqueadores de la IL-1 han aparecido como la alternativa terapéutica de elección en estos síndromes. En este sentido, se ha demostrado que anakinra, la forma recombinante humana del antagonista del receptor de IL-1 (IL-1Ra), genera excelentes respuestas tanto clínicas como bioquímicas en estos pacientes13-17. En la actualidad, diversas compañías farmacéuticas están desarrollando diferentes compuestos bloqueadores de la IL-1 que pueden ser abordajes terapéuticos alternativos para estos pacientes en un futuro próximo18.
GRANULOMATOSIS SISTÉMICAS PEDIÁTRICAS: SÍNDROME DE BLAU Y SARCOIDOSIS DE INICIO PRECOZEste subgrupo de enfermedades autoinflamatorias hereditarias está compuesto por el síndrome de Blau (MIM 186580) y la sarcoidosis de inicio precoz (MIM 609464)19-21. Si bien fueron descritas como entidades clínicas independientes, en la actualidad se las considera una misma enfermedad, con una misma base genética y un mecanismo etiopatogénico común. La diferencia entre una y otra estriba en que haya o no historia familiar de la enfermedad. Así, el término síndrome de Blau se emplea para designar a los pacientes con antecedentes familiares, mientras el término sarcoidosis de inicio precoz se reserva para los casos esporádicos, sin historia familiar de la enfermedad22.
Desde un punto de vista clínico, las granulomatosis sistémicas pediátricas se caracterizan por un curso inflamatorio persistente, con afección principal de la piel, las articulaciones y los ojos23. A pesar del curso persisenfermedad, en episodios agudos, sin que hayan sido identificados factores que los desencadenen. La edad de inicio de la enfermedad se sitúa habitualmente por debajo de los 4 años, y las manifestaciones cutáneas y/o articulares son las primeras en detectarse, aumentando de forma progresiva el cortejo sintomático conforme evoluciona la enfermedad. La afección cutánea consiste en un exantema eritematoso generalizado, discretamente granular por la presencia de granulomas no caseificantes a nivel dérmico, presente habitualmente desde el inicio de la enfermedad. Las manifestaciones articulares también son habituales en el inicio de la enfermedad; la forma de afección articular más frecuente es una artritis poliarticular crónica, simétrica, que afecta a grandes y pequeñas articulaciones y se acompaña de una tenosinovitis intensa, no dolorosa, debida al infiltrado granulomatoso de la sinovia, que puede ocasionar una limitación del movimiento articular y obliga a la realización de sinovectomías en algún momento de la vida de estos pacientes. Las manifestaciones oculares constituyen el tercer gran grupo de manifestaciones clínicas, y son las que originan mayor morbilidad. Son poco frecuentes en el inicio de la enfermedad, y asimismo son muy raras como manifestaciones únicas. La forma más frecuente de afección ocular es la uveítis, que puede ser anterior, posterior o más comúnmente panuveítis, granulomatosa o no granulomatosa, pudiendo afectar a uno o a ambos globos oculares24,25.
Además de la típica tríada de afección cutánea, articular y ocular, se han descrito diferentes manifestaciones menos frecuentes, tales como: fiebre, neuropatías craneales transitorias, afección renal granulomatosa, afección granulomatosa hepática, afección cardíaca, tanto en la forma de miocarditis granulomatosa como cardiopatía secundaria al desarrollo de hipertensión maligna, y adenopatías23,26-30.
El hallazgo más específico de este grupo de enfermedades es la presencia de múltiples granulomas no caseificantes en los tejidos y órganos afectos, que no se deben a ningún agente infeccioso (micobacterias, brucella, etc.) y son semejantes a los observados en la sarcoidosis del adulto23. No obstante, la edad de inicio tan temprana, las principales manifestaciones clínicas y la ausencia de manifestaciones pulmonares en estos pacientes permiten establecer un diagnóstico diferencial con la sarcoidosis del adulto31.
EtiopatogeniaDesde el punto de vista genético, las granulomatosis sistémicas pediátricas presentan un patrón de herencia autosómica dominante. En 2001 se identificó el gen CARD15 (actualmente conocido como NOD2) como causante del síndrome de Blau32. Dicho gen codifica para la proteína Nod2, que forma parte de la familia de receptores NLR comentada anteriormente al hablar de los síndromes CAPS, y que intervienen en la respuesta inmunitaria innata33. En 2005 dos grupos independientes identificaron mutaciones en el gen CARD15 en pacientes afectos de sarcoidosis de inicio precoz, y algunas de ellas son las mismas que se había identificado como causa del síndrome de Blau34,35. De esta manera se estableció un mecanismo etiopatogénico común para ambas entidades y se zanjó la histórica discusión referente a si ambas entidades eran una misma enfermedad o eran enfermedades diferentes36.
TratamientoEn lo referente a los tratamientos, no se han observado respuestas clínicas positivas a colchicina, AINE ni a muchos de los fármacos antirreumáticos de uso más extendido. Muchas de las manifestaciones sistémicas pueden mejorar o desaparecer con el empleo de corticoides a dosis altas, pero su uso prolongado tiene importantes efectos secundarios en pacientes de edad pediátrica. En la actualidad, los agentes biológicos bloqueadores del factor de necrosis tumoral (TNF) se presentan como alternativas terapéuticas para controlar la enfermedad y mejorar, si no todas, parte de las manifestaciones clínicas.
SÍNDROME DE ARTRITIS PIÓGENA ESTÉRIL, PIODERMA GANGRENOSO Y ACNÉ (PAPA)La enfermedad autoinflamatoria conocida como síndrome PAPA (Pyogenic sterile Arthritis, Pyoderma gangrenosum and Acne [MIM 604416]) fue descrita por vez primera en 1997, en el seno de una familia con un claro patrón de herencia autosómico dominante; con posterioridad se han descrito diferentes familias afectas del mismo cuadro37,38. Desde un punto de vista clínico, las manifestaciones inflamatorias se inician a edad temprana, antes de los 10 años de edad, y afectan fundamentalmente a las articulaciones y la piel.
La forma habitual de afección articular es una artritis intermitente, migratoria, destructiva, monoarticular u oligoarticular, que afecta a grandes articulaciones y tiene como característica principal un intenso infiltrado polimorfonuclear que confiere al liquido sinovial un aspecto purulento, pero estéril.
Las manifestaciones cutáneas suelen presentarse más tardíamente que las manifestaciones articulares, y se pueden diferenciar diferentes tipos, que se inician asimismo en momentos diferentes y perduran toda la vida. El pioderma gangrenoso es la primera manifestación cutánea en aparecer, habitualmente antes de los 10 años de edad. Se caracteriza por múltiples lesiones papulares eritematosas, ligeramente quísticas, localizadas preferentemente en las extremidades inferiores, y suelen aparecer tras traumatismos menores. Conforme evolucionan, estas lesiones tienden a ulcerarse, cronificándose y llegando a estar presentes durante largos periodos. Abscesos estériles en los puntos de inyección, tanto intradérmica como parenteral, lo cual indica un fenómeno de patergia.
A partir de la pubertad y la adolescencia aparece un acné quístico grave que afecta a áreas corporales extensas y perdura durante la edad adulta; se caracteriza por el desarrollo de lesiones quísticas, deformantes, causa de los trastornos psicológicos y sociales que padecen los pacientes afectos del síndrome PAPA.
A semejanza de lo que se ha comentado para otras enfermedades autoinflamatorias hereditarias, no existen parámetros de laboratorio específicos del síndrome PAPA. Así, durante las fases activas de la enfermedad, se puede identificar leucocitosis debida a neutrofilia, trombocitosis y una intensa reacción de fase aguda, con un incremento de la VSG y de los valores plasmáticos de los reactantes de fase aguda. Asimismo, no se detectan microorganismos causantes de las manifestaciones articulares o cutáneas, como tampoco evidencias de enfermedad autoinmunitaria.
EtiopatogeniaEn 2001 se identificó la base molecular del síndrome PAPA, al identificarse mutaciones que segregan con la enfermedad en el gen CD2BP1, localizado en el cromosoma 15q22-2439. Dicho gen codifica la proteína PSTPIP1 (del inglés proline serine threonine phosphatase-interacting protein 1), y hasta el momento actual se han identificado tres mutaciones diferentes asociadas a la enfermedad (Ala-230-Thr, Glu-250-Gln y Asp-266-Asn), localizadas todas ellas en la misma región funcional de la proteína.
Se conoce que el gen CD2BP1 se expresa fundamentalmente en el pulmón y en monocitos y neutrófilos, pero hay pocos datos referentes a la función o funciones de la proteína codificada por él. Así, entre las funciones bien establecidas de dicha proteína se encuentra su papel en la reorganización de la actina del citoesquele-40,41. Recientemente, como consecuencia de los estudios llevados a cabo en el síndrome PAPA, se ha propuesto un posible papel regulador de la actividad general del inflamasoma, mediante la interacción física con la proteína pirina/marenostrina, causa de la fiebre mediterránea familiar42. En este sentido, se ha demostrado una producción incrementada de la citocina proinflamatoria IL-1β en los pacientes afectos del síndrome PAPA, así como una excelente respuesta clínica de dichos pacientes al tratamiento con el agente bloqueador de IL-1 anakinra42,43.
TratamientoEn lo referente al tratamiento farmacológico, se han probado múltiples abordajes (AINE, corticoides sistémicos, corticoides intraarticulares, antibióticos, metotrexato), que no han generado respuestas clínicas positivas prolongadas y obligan a que los pacientes sean sometidos a múltiples drenajes articulares para extraer las colecciones purulentas. El pronóstico de estos pacientes ha mejorado en los últimos años, con las buenas respuestas clínicas observadas tanto con agentes biológicos anti-TNF (etanercept, infliximab), así como con bloqueadores de la IL-1 (kineret)43-45.
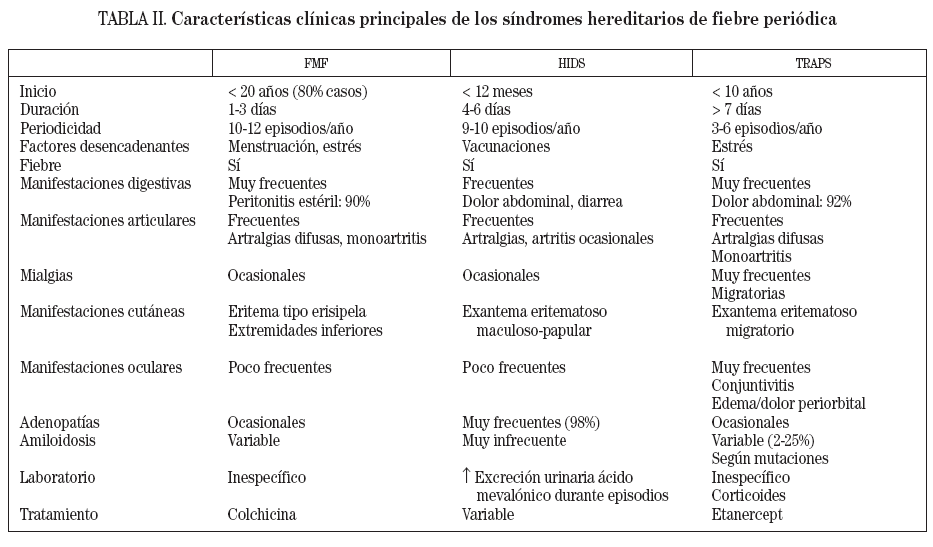
SÍNDROMES HEREDITARIOS DE FIEBRE PERIÓDICALos síndromes hereditarios de fiebre periódica constituyen el subgrupo más importante entre las enfermedades autoinflamatorias. Agrupa dos enfermedades con un patrón de herencia autosómico recesivo, la fiebre mediterránea familiar (FMF) y el síndrome de hiper-IgD y fiebre periódica (HIDS), y una entidad con herencia autosómica dominante, el síndrome periódico asociado al receptor del TNF (TRAPS)3. Todos estos síndromes presentan episodios inflamatorios agudos, autolimitados, de duración variable, que recurren de manera periódica. En la tabla II se resumen las principales características clínicas y genéticas de estos síndromes. Debido a que sólo el síndrome TRAPS presenta un importante componente de afección cutánea, será el único síndrome hereditario de fiebre periódica comentado en detalle en la presente revisión, y emplazamos al lector interesado al apartado de bibliografía para localizar revisiones o artículos sobre el tema.
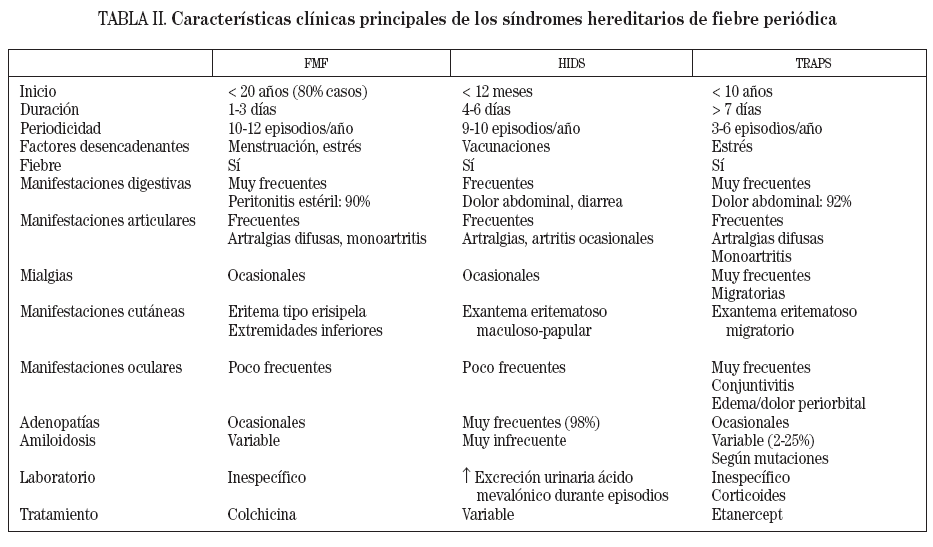
TABLA II. Características clínicas principales de los síndromes hereditarios de fiebre periódica
SINDROME PERIÓDICO ASOCIADO AL RECEPTOR DEL TNF (TRAPS)Las primeras descripciones clínicas de un síndrome de fiebre periódica con herencia dominante, conocido en la actualidad como síndrome TRAPS, datan de los años ochenta, cuando se dieron a conocer diferentes familias afectas aparentemente por la misma enfermedad. En aquel momento se dieron nombres diversos tales como fiebre hiberniana familiar o síndrome de fiebre periódica autosómica dominante con amiloidosis46-48. A raíz del descubrimiento de su base molecular en 1999, se acordó sustituir los diferentes nombres por el acrónimo TRAPS (del inglés TNF receptorassociated periodic syndrome [MIM 142680]) para definir un síndrome de fiebre periódica, de herencia dominante, debido a mutaciones en el receptor 1 del TNF1.
Los pacientes afectos de TRAPS presentan episodios inflamatorios agudos, de duración prolongada (más de 7 días, pudiendo alcanzar las 3-4 semanas de duración), que comienzan antes de los 10 años de edad49. Clínica-mente, hay una serie de síntomas presentes en la mayoría de los episodios, como fiebre y manifestaciones osteomusculares, cutáneas, oculares y digestivas.
La fiebre se observa en la gran mayoría de los pacientes y suele alcanzar los 40-41 °C. No se ha observado un patrón febril típico.
Las manifestaciones reumáticas y las cutáneas son los rasgos clínicos distintivos del síndrome TRAPS, presentes en la práctica totalidad de los pacientes. Dichas manifestaciones se caracterizan por la aparición de intensas mialgias en grupos musculares aislados, migratorias en sentido centrífugo, debidas a una fascitis monocítica inflamatoria, acompañadas por un exantema eritematoso, habitualmente maculopapular, caliente y doloroso a la palpación, que afecta a las áreas cutáneas situadas por encima de los grupos musculares afectados. Asimismo son frecuentes las poliartralgias, y relativamente poco frecuentes las artritis, que tienden a ser monoarticulares u oligoarticulares.
Las manifestaciones oculares están presentes en más del 80% de los pacientes, habitualmente como conjuntivitis, edema periorbital y dolor periorbital. Es infrecuente la aparición de uveítis o papiledema.
Las manifestaciones digestivas están presentes hasta en el 92% de los pacientes, habitualmente como dolor abdominal secundario a una peritonitis inflamatoria o a una inflamación de los músculos de la pared abdominal.
A semejanza de muchas enfermedades autoinflamatorias, la complicación más grave del síndrome TRAPS es la aparición de amiloidosis secundaria, habitualmente renal, como consecuencia de procesos inflamatorios repetidos a lo largo de años49.
EtiopatogeniaLas bases moleculares del síndrome TRAPS fueron establecidas en 1999, al identificarse mutaciones que segregaban con la enfermedad en el gen TNFRSF1A, que codifica para el receptor 1 del TNF (también denominado p55 y CD120a)1. Desde entonces, se han identificado más de 60 mutaciones que causan enfermedad, la mayoría de ellas localizadas en los exones 2, 3 y 4 del gen.
No existe un mecanismo fisiopatológico claro que explique por qué mutaciones en el gen TNFRSF1A dan lugar a un síndrome de fiebre periódica. Se han propuesto diferentes modelos, todos ellos pendientes de demostración experimental. El modelo con una mayor aceptación propone un defecto en el shedding del receptor como base de la enfermedad1. Según este modelo, las mutaciones del gen provocarían cambios en la proteína que afectan al shedding o degradación proteinolítica de los dominios extracelulares del receptor, que son el mecanismo homeostático natural de control de la señal inflamatoria a través de este receptor.
TratamientoDesde un punto de vista terapéutico, los pacientes afectos del síndrome TRAPS muestran ausencia de respuesta a colchicina y AINE, pero tienen buena respuesta a los corticoides a dosis altas49. En función de la severidad de los síntomas y de la edad de los pacientes, el abordaje terapéutico con corticoides puede irse modulando de unos pacientes a otros. Así, en pacientes pediátricos se recomienda el empleo puntual de corticoides para cortar los episodios inflamatorios agudos, mientras que en pacientes adultos o pacientes pediátricos no respondedores a corticoides se recomienda el empleo del agente biológico anti-TNF etanercept. Debido a la posible disociación entre la respuesta clínica (habitualmente buena o muy buena) y la respuesta bioquímica (parámetros bioquímicos de inflamación en valores altos con respecto a los considerados normales), se recomienda un estrecho seguimiento de dichos parámetros bioquímicos, así como de la función renal, digestiva o tiroidea en estos pacientes, para evaluar el riesgo de desarrollo de amiloidosis secundaria50-52. Finalmente, la buena respuesta clínica y bioquímica observada en unos pocos pacientes de TRAPS tratados con el agente bloqueador de la IL-1 anakinra abre la posibilidad de su uso terapéutico en el síndrome TRAPS53.
Correspondencia: Dr. J.I. Aróstegui. Unidad de Enfermedades Autoinflamatorias. Servicio de Inmunología. Centro de Diagnóstico Biomédico (CDB). Hospital Clínic. Villarroel, 170. 08036 Barcelona. España. Correo electrónico: jiaroste@clinic.ub.es