

















- Las lentiginosis se caracterizan por un número excepcionalmente alto de lentigos que se distribuyen de forma característica.
- Los lentigos acostumbran ser el primer signo de las lentiginosis sistémicas, muchas de ellas familiares y, por lo general, con un patrón de herencia autosómica dominante.
- Ante una lentiginosis se debe descartar siempre alteraciones extracutáneas, sobre todo cardíacas e intestinales, y valorar el riesgo de neoplasias.
- Los factores condicionantes del pronóstico de las principales lentiginosis sistémicas son la afección cardíaca en el síndrome LEOPARD y en el complejo de Carney y el riesgo de neoplasia maligna en el síndrome de Peutz-Jeghers.
- En la actualidad se conocen algunas de las mutaciones genéticas que causan algunas lentiginosis como el síndrome LEOPARD, el síndrome de Peutz-Jeghers y el complejo de Carney.
Los lentigos son hipermelanosis epidérmicas benignas debidas a un aumento en el número de melanocitos y de la producción de la melanina. Clínicamente se manifiestan como pequeñas máculas hiperpigmentadas, de color marrón a negro generalmente uniforme, bien delimitadas, por lo general de diámetro < 5 mm y localización variable. Se distinguen dos tipos fundamentales de lentigos: los lentigos simples, que pueden aparecer en cualquier superficie mucocutánea (fig. 1), y los lentigos solares, actínicos o seniles, que se desarrollan exclusivamente en áreas fotoexpuestas inducidos por las radiaciones ultravioletas (fig. 2). Histológicamente, los lentigos presentan hiperpigmentación de la capa basal epidérmica con proliferación de melanocitos, acantosis y elongación regular de las crestas interpapilares epidérmicas (fig. 3). Ocasionalmente puede observarse incontinencia pigmentaria con melanófagos en la dermis papilar (fig. 4). Los lentigos deben diferenciarse de las efélides o pecas que aparecen en zonas fotoexpuestas, y presentan un aumento de la cantidad de melanina pero no del número de melanocitos (fig. 5).
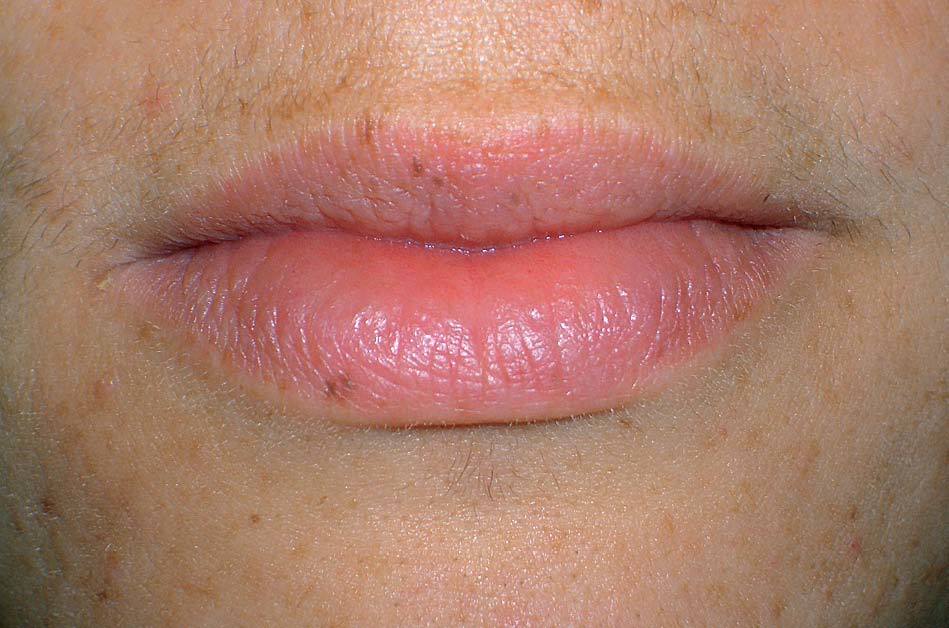
Figura 1. Lentigos simples en los labios.
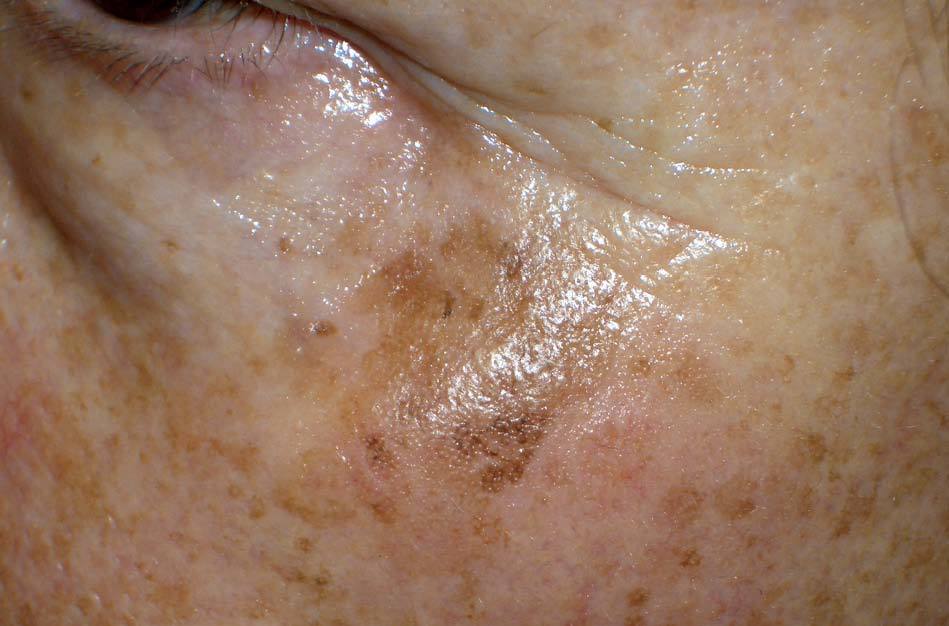
Figura 2. Lentigos solares.
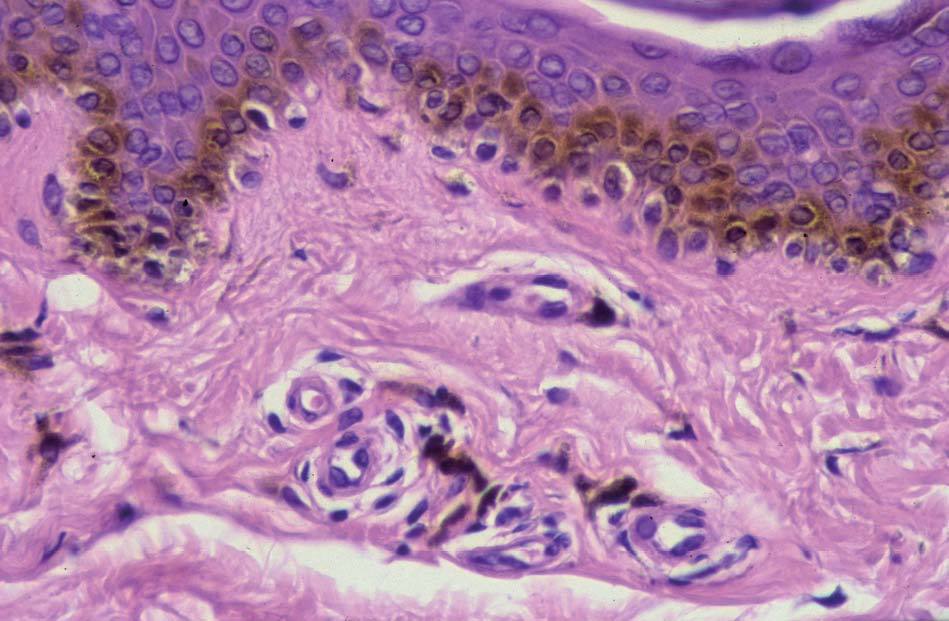
Figura 3. Hiperpigmentación de la capa basal epidérmica con proliferación de melanocitos.
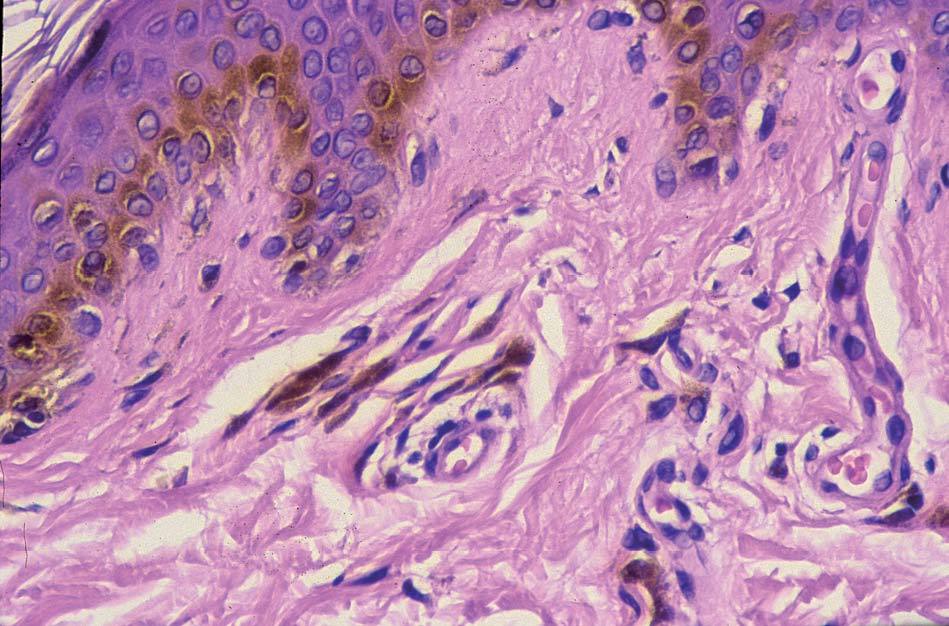
Figura 4. Melanófagos en la dermis papilar.
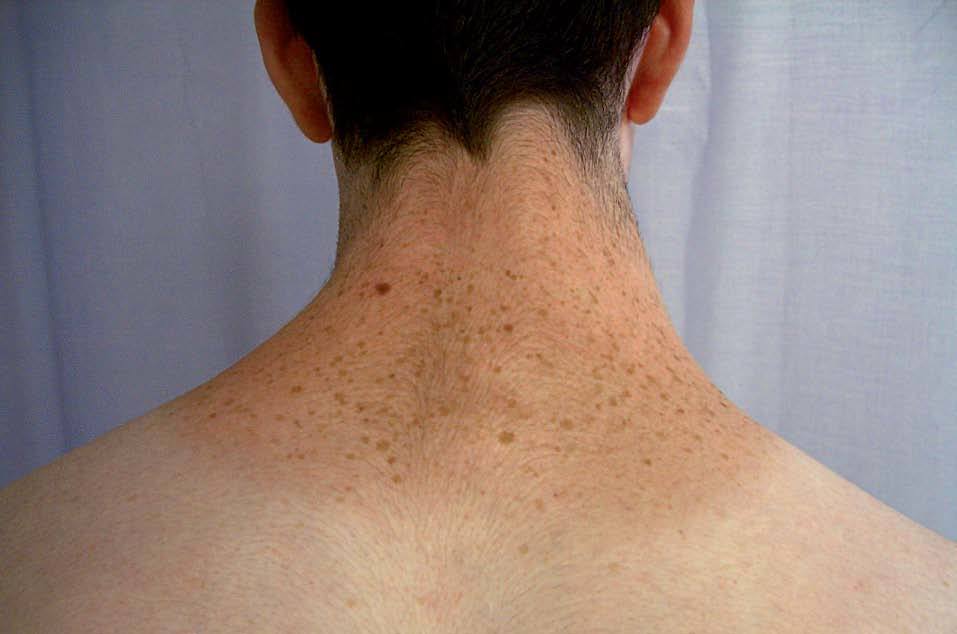
Figura 5. Efélides o pecas.
Las lentiginosis son un grupo heterogéneo de cuadros o síndromes, congénitos o adquiridos, caracterizados por la aparición de una cantidad excepcionalmente alta de lentigos simples, por lo general en edades tempranas de la vida. Las lentiginosis pueden clasificarse en función de la distribución de los lentigos en: lentiginosis múltiples, con lentigos distribuidos de manera generalizada, y lentiginosis circunscritas, con lentigos limitados a un área o región. A su vez, estas lentiginosis se subdividen en dos grupos: lentiginosis cutaneomucosas aisladas, sin trastornos extracutáneos asociados, y lentiginosis sistémicas, asociadas a alteraciones extracutáneas características como los síndromes cardiocutáneos y las lentiginosis asociadas a poliposis digestivas (tabla I). En las lentiginosis sistémicas, el lentigo puede considerarse una proliferación hamartomatosa de melanocitos que se asocia a hiperplasias, hamartomas y neoplasias en otros tejidos, sobre todo de origen mesenquimal y endocrino. Se cree que el origen de estos síndromes es un defecto genético precoz en el desarrollo embrionario que afecta al neuroectodermo y/o el mesodermo1 o en diferentes períodos del desarrollo individual2. La mayoría de las lentiginosis sistémicas corresponden a procesos hereditarios transmitidos con un patrón de herencia autosómica dominante con expresividad variable dentro de la misma familia. Los lentigos acostumbran ser el primer signo de estos síndromes y el resto de alteraciones suelen desarrollarse en edades medias de la vida. Las lentiginosis familiares cumplen los criterios de los síndromes familiares neoplásicos: la enfermedad es más severa en los casos esporádicos que en los familiares, diferentes generaciones de una misma familia se hallan afectas de diferentes tipos de tumores, frecuentemente bilaterales, y presentan el fenómeno de anticipación.

TABLA I. Clasificación de las lentiginosis
La identificación en los últimos años de alteraciones cromosómicas específicas de algunas lentiginosis permite diferenciar cuadros que han sido confundidos en la literatura a lo largo de décadas, además de predecir el desarrollo de anomalías extracutáneas y neoplasias malignas, realizar el consejo genético familiar y diseñar protocolos de manejo.
LENTIGINOSIS SIN TRASTORNOS SISTÉMICOSMúltiplesLentiginosis generalizada, profusa o difusa. Se caracteriza por la aparición de múltiples lentigos en la infancia, por separado o en pequeños brotes a intervalos irregulares, en toda la superficie cutánea excepto las mucosas3. Algunos autores la consideran una forma menor del síndrome LEOPARD que no asocia trastornos sistémicos4. Su patogenia es desconocida, pero algunos casos se han relacionado con mutaciones en el cromo-soma 4, en 4q21.1-q22.35 y específicamente del gen c-kit6. Se ha descrito su asociación ocasional en casos familiares con estrabismo7 y otros trastornos pigmentarios como máculas café con leche, máculas hipopigmentadas lanceoladas e hipopigmentación en confetti8.
Lentiginosis eruptiva. Se caracteriza por la aparición brusca de múltiples lentigos que persisten semanas o meses en adolescentes y adultos jóvenes. Las lesiones pueden adoptar inicialmente un aspecto telangiectásico, pero rápidamente evolucionan a máculas pigmentadas que pueden corresponderse a lentigos o nevos melanocíticos celulares. También se han descrito puntualmente casos familiares en que no se conoce hasta el momento una causa genética9.
CircunscritasLentiginosis unilateral parcial (LUP), segmentaria, agminada o zosteriforme. Es un proceso poco frecuente que afecta habitualmente a mujeres caucásicas. La LUP se caracteriza por la aparición de numerosos lentigos de color marrón claro, de 2-10 mm de diámetro, sobre piel normal, agrupados en un segmento del cuerpo y con un borde bien definido limitado en la línea media (fig. 6). Aunque puede estar presente ya en el nacimiento, acostumbra aparecer entre los 5 y los 15 años de edad, con una discreta progresión en los meses o años posteriores. La LUP suele localizarse en el tronco, la cara, el cuello y las extremidades superiores10, habitualmente en un único dermatoma y con mayor frecuencia en el hemicuerpo izquierdo11. Sin embargo, se han descrito patrones de distribución segmentarios, metaméricos, curvilíneos y arremolinados, así como afección por varios dermatomas no siempre contiguos12, la afección segmentaria bilateral y la alternancia de varios segmentos afectados. Histológicamente, se puede observar, además del patrón de lentigo, un patrón de «jentigo» con pequeños nidos de melanocitos en la unión dermoepidérmica. Se han observado otros trastornos cutáneos asociados a la LUP, como fragilidad cutánea, xerosis difusa con áreas hiperqueratósicas, pezón supernumerario, nevo vascular, nevo azul13, nevo acrómico, cutis marmorata, acantosis nigricans y vitíligo14. Algunos casos asociados a máculas café con leche, efélides axila-res, nódulos de Lisch y neurofibromas segmentarios homolaterales, contralaterales o bilaterales respecto la LUP han sido considerados formas peculiares de neurofibromatosis segmentaria15,16. Se desconoce su patogenia, pero se ha propuesto que se trata de un mosaicismo somático de los melanoblastos de la cresta neural. También se ha señalado que la LUP podría ser una expresión incompleta de un síndrome lentiginoso sistémico, pues se han descrito casos asociados a enfermedad celíaca, asma14 y trastornos cardíacos, neurológicos13, reumáticos, endocrinos17 y hemáticos18.
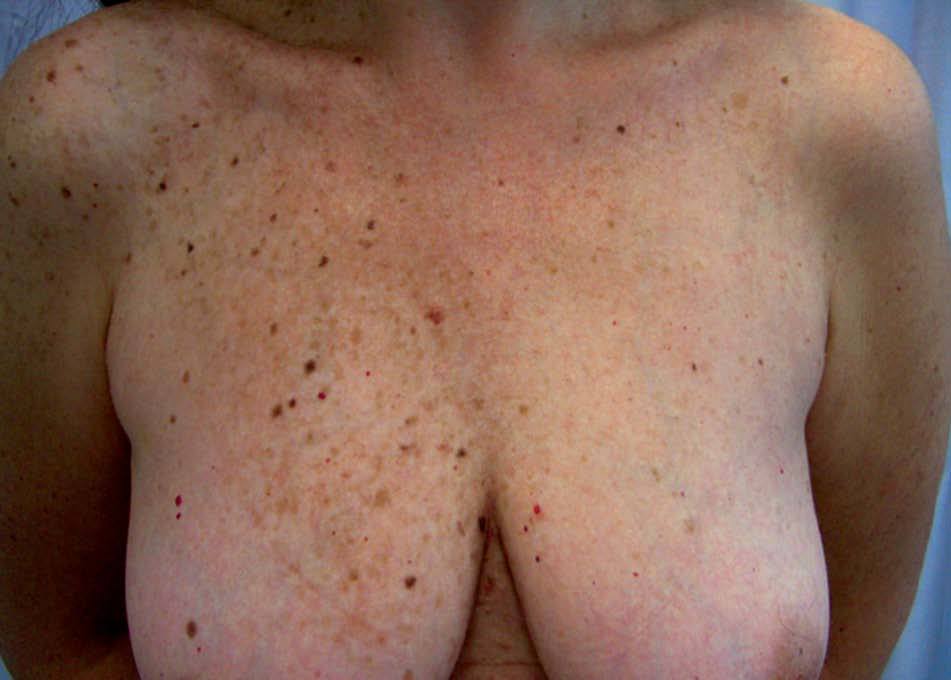
Figura 6. Lentiginosis unilateral parcial o segmentaria.
Síndrome de Laugier-Hunziker. Es un trastorno benigno adquirido caracterizado por la aparición de hiperpigmentación macular en los labios y en la mucosa oral19. Es más frecuente en mujeres en la edad adulta y raramente se ha observado que remita. La mayoría de los casos son esporádicos, pero se han descrito excepcionalmente casos familiares20, de los que no se conoce aún causa genética.
Las lesiones pigmentadas pueden variar entre marrón, gris o negro y pueden ser aisladas o confluentes. La hiperpigmentación en los labios afecta más frecuentemente al inferior y puede adoptar una forma lineal. La pigmentación oral afecta sobre todo a la mucosa bucal y el paladar duro, pero también puede aparecer en las comisuras bucales, las encías, el suelo de la boca y la lengua.
En el 60% de los casos se afectan las uñas y otras superficies cutáneas como las del cuello, el tórax, el abdomen, las yemas de los dedos, las palmas, las plantas y más raramente, los genitales, el periné y la región perianal. La manifestación ungueal más común es una melanoniquia longitudinal marrón-negra de 1-2 mm de grosor, que ocasionalmente puede evidenciar un signo de Hutchinson21 y debe diferenciarse de la melanoniquia estriada. Otras manifestaciones ungueales son la doble banda en los laterales de la lámina ungueal y la pigmentación homogénea en la mitad radial de la uña22. También se ha descrito la afección concomitante de otras mucosas como la conjuntiva y el esófago.
Lentiginosis hereditaria de la raza negra. Esta lentiginosis, descrita por O’Neill en 10 miembros adultos de una familia de raza negra con un patrón de herencia dominante, se caracteriza por la aparición de múltiples lentigos durante la lactancia o la primera infancia en el área centrofacial23. También pueden afectarse los labios, los glúteos, las extremidades y las superficies palmoplantares. No se han observado lesiones mucosas. Se ha descrito un caso similar en un varón caucásico con un tío materno afecto2. Hasta ahora se desconoce su patogenia y no se ha identificado el gen que lo causa.
Otras lentiginosis circunscritas. Entre ellas, la lentiginosis perigenitoaxilar (con lentigos limitados en las zonas de secreción apocrina, como periné y axilas); la lentiginosis perianal24 y la lentiginosis blanca25 (un proceso congénito caracterizado por máculas hiperpigmentadas «en gotas» en las áreas fotoexpuestas e hipomelanóticas en las zonas cubiertas).
LENTIGINOSIS MÚLTIPLES CON TRASTORNOS SISTÉMICOSSíndrome LEOPARD o de los lentigos múltiples, lentiginosis cardiomiopáticaEs un síndrome cardiocutáneo hereditario poco frecuente, transmitido con un patrón autosómico dominante de alta penetrancia, ligeramente más frecuente en varones, caracterizado por la asociación de lentiginosis y un grupo heterogéneo de alteraciones del desarrollo (tabla II). El nombre del síndrome corresponde al acrónimo en inglés de sus componentes más comunes: Lentigos múltiples, defectos de la conducción en el Electrocardiograma, hipertelorismo Ocular, estenosis Pulmonar, genitales Anormales, Retraso del crecimiento y sordera neurosensitiva (Deafness).

TABLA II. Anomalías asociadas al síndrome LEOPARD
La lentiginosis suele ser la manifestación clínica inicial y la más frecuente en este síndrome. Las lesiones son máculas marrón negruzco, irregulares, de tamaño variable entre puntiforme y 5 mm, presentes en el nacimiento o de aparición durante los primeros años de vida, con una tendencia a aumentar en número hasta la pubertad. Suelen distribuirse por todo el cuerpo excepto las mucosas, pero son más numerosos en el cuello, la cara y la parte superior del tronco (fig. 7). Generalmente son lesiones estables, pero se ha descrito su involución en edades medias de la vida, en ocasiones dejando máculas hipopigmentadas residuales.
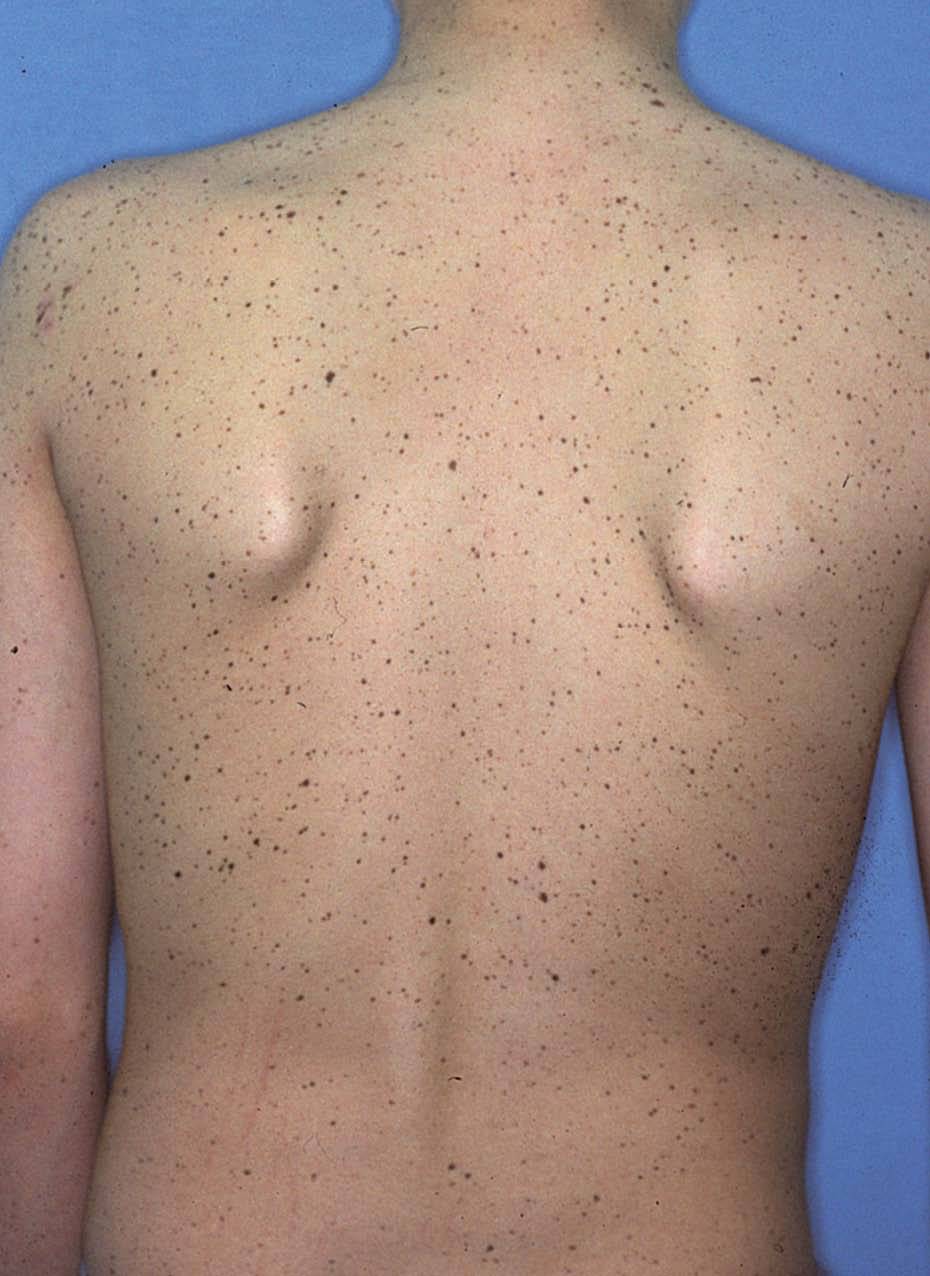
Figura 7. Síndrome LEOPARD: lentigos característicos en la espalda de un adolescente.
La afección cardíaca es frecuente en el síndrome LEOPARD y es la principal causa de morbimortalidad incluso en edades tempranas. La afección cardíaca más frecuente es las alteraciones de la conducción, aunque también son comunes las malformaciones anatómicas como la estenosis pulmonar (40%) y la miocardiopatíahipertrófica primaria. Éstas presentan instauración y progresión variables, por lo que se recomiendan evaluaciones periódicas cada 6 meses con un electrocardiograma, una radiografía de tórax y una ecocardiografía. Las alteraciones dismórficas cefálicas como el hipertelorismo ocular (25%) y el prognatismo mandibular condicionan unos rasgos faciales característicos del síndrome LEOPARD. La sordera neurosensitiva, que acostumbra manifestarse hacia los 9 meses de vida, y las anomalías genitales (26%) son hallazgos menos constantes en este síndrome. Los trastornos genitales predominan en los varones y los más frecuentes son la hipoplasia gonadal y la hipospadias. Se han propuesto unos criterios mínimos para el diagnóstico de síndrome LEOPARD7: a) múltiples lentigos junto con al menos dos de las otras características del síndrome, o b) en ausencia de lentigos cutáneos, como mínimo tres de las otras características del síndrome y tener un familiar directo diagnosticado de síndrome LEOPARD según los criterios de a). La mayoría de los pacientes presentan de 3 a 5 de las características diagnósticas y algunas de ellas no se manifiestan hasta la pubertad o más tarde. Ante la ausencia de lentigos en la infancia, puede sospecharse de síndrome LEOPARD en los primeros meses de vida si se evidencian las otras tres características principales de este síndrome, como los rasgos faciales, la miocardiopatía hipertrófica y las máculas café con leche26.
El síndrome LEOPARD guarda similitudes con el síndrome de Watson (que combina lentigos, máculas café con leche, estenosis pulmonar, defectos cardíacos, retraso mental y sordera27), el síndrome de Moynahan (con múltiples nevos en las articulaciones, hipoplasia genital, testes ectópicos, estenosis mitral congénita, retraso del crecimiento e infantilismo psíquico28) y la enfermedad de Dowling-Degos (con lentigos, lesiones pigmentadas periorales, quistes epidermoides múltiples y senos pilonidales). Se ha descrito asimismo la asociación del síndrome LEOPARD con la tétrada de Gerstmann, el síndrome de Noonan (con o sin neurofibromatosis), el síndrome de Werner, el síndrome de Marfan, la distrofia elástica, la sinovitis vellonodular pigmentada, la depresión y la psicosis familiar.
La patogenia del síndrome LEOPARD es desconocida. Se han propuesto dos teorías al respecto: a) un defecto de la cresta neural daría lugar a una hiperactividad melanocitaria cutánea y a un aumento de la acción de factores alfaadrenérgicos sobre el músculo cardíaco que causarían la miocardiopatía y las alteraciones en el electrocardiograma, y b) un defecto genético en el neurectodermo, donde las líneas celulares anormales de la cresta neural interaccionan con las células del mesodermo, daría lugar a anomalías cutáneas, neurológicas y cardíacas4. Esta última teoría explicaría la asociación del síndrome LEOPARD con la neurofibromatosis, la esclerosis tuberosa y el feocromocitoma25. Se han descrito diferentes mutaciones que podrían explicar los diferentes grados de expresión dentro de una misma familia. El 88% de los pacientes con LEOPARD presentan alteraciones alélicas en el gen PTPN11 de la neurofibromatosis I, en el locus 12q24.128: este gen codifica la proteína tirosinfosfatasa SPH-2, con propiedades angiogénicas y mitogénicas en el músculo liso vascular. Algunos autores han hallado correspondencia entre la mutación en PTPN11 y la miocardiopatía hipertrófica29. El diagnóstico del síndrome LEOPARD debe confirmarse con el estudio de las mutaciones del gen PTPN1130 .
Complejo de Carney o síndrome de los mixomas múltiplesEl complejo de Carney (CC) se caracteriza por el desarrollo de lesiones tumorales en los tejidos mesenquimales, endocrinos y derivados de la cresta neural. Combina lesiones cutáneas pigmentadas polimorfas, mixomas cardíacos y mucocutáneos, schwannomas e hiperactividad endocrina. En el CC se han incluido otros dos síndromes: el síndrome LAMB (Lentigo, mixoma Auricular, mixomas Mucocutáneos y nevo azul [Blue]) y el síndrome NAME (Nevos azules, mixomas Auriculares, neurofibromas Mixoides mucocutáneos o mucinosis cutánea y Efélides o hiperactividad endocrina). El CC es un proceso poco frecuente del que se han descrito más de 150 casos, que en un 50% se puede considerar familiares31.
Las lesiones pigmentadas mucocutáneas son la característica más frecuente del CC y acostumbran ser la primera manifestación en edades jóvenes. Corresponden tanto a lentigos y nevos compuestos como a nevos azules. Hasta un 10% de los pacientes tienen ambos tipos de lesiones. Los lentigos son similares a los observados en el síndrome LEOPARD: pequeñas máculas de 0,2-2 mm, marrón negruzco, redondas o irregulares, planas o levemente elevadas, distribuidas predominantemente en el área centrofacial (alrededor de la boca, el puente nasal, los párpados y alrededor de los ojos) y el borde del bermellón. Otras localizaciones características de los lentigos en el CC son los pabellones auriculares, el cuello, el tronco superior, las extremidades y el dorso de las manos. También pueden hallarse lesiones mucosas en la conjuntiva (característicamente en la carúncula lagrimal y el pliegue semilunar), la esclera, la mucosa oral y la vulva, sobre todo en los labios menores. Con menor frecuencia aparecen lentigos en las palmas y las plantas, la región perianal, el cuero cabelludo, los pulpejos digitales o el glande. Los nevos azules son pápulas negroazuladas, de hasta 8 mm de diámetro, habitualmente únicas, localizadas en la cara, el tronco y las extremidades, excepto las manos y los pies. También pueden aparecer en el CC efélides y nevos junturales.
Los mixomas están presentes en el 60% de casos con CC y pueden aparecer, por orden de frecuencia, en el corazón, la piel, las mucosas y las mamas. Suelen ser múltiples y tienen tendencia a recurrir después de su exéresis. Los mixomas mucocutáneos (33%) pueden aparecer desde el nacimiento hasta la cuarta década de la vida, y su presencia obliga a descartar un mixoma cardíaco oculto. Acostumbran ser lesiones asintomáticas en forma de pápulas sésiles o nódulos subcutáneos de superficie lisa, habitualmente de menos de 1 cm y coloración blanquecina o de la piel normal, que en ocasiones pueden confundirse con epiteliomas basocelulares. Pueden presentarse agrupados y las localizaciones más frecuentes son la cabeza, el cuello y el tronco, aunque característicamente afecta a los párpados, el canal auditivo externo y los pezones. También pueden aparecer en genitales, glúteos, ingles, extremidades (brazos, axilas, hombros, piernas, excepto manos y pies), cavidad oral (paladar, lengua), conjuntiva y esclera. Histológicamente son tumores sólidos bien delimitados en dermis y/o sub-cutis, en ocasiones con zonas quísticas, y están compuestos por células mesenquimales de aspecto similar a los fibroblastos, matriz mucinosa, capilares, fibras de colágeno y reticulina, mastocitos y células inflamatorias.
Los mixomas cardíacos están presentes en más del 65% de los pacientes con CC y son su componente más grave, pues causa el 50% de la mortalidad por CC. En el 50% de los casos son múltiples y pueden afectar a una o varias cámaras, más frecuentemente la aurícula izquierda, por lo que presentan un comportamiento más agresivo que los mixomas cardíacos esporádicos. Suelen detectarse en la infancia o la adolescencia y en un 50% de los casos están presentes en el momento de la consulta junto con las lesiones pigmentadas, pero pueden ser la primera manifestación en pacientes con menor número de lentigos faciales.
Hasta el 25% de las mujeres con CC presentan alteraciones mamarias, la mayoría benignas, como fibradenomas mixoides mamarios asintomáticos, habitualmente múltiples y bilaterales.
Las manifestaciones de hiperactividad endocrina en el CC son numerosas y se deben fundamentalmente a alteraciones adrenocorticales y testiculares. La más frecuente (20-30%) es un síndrome de Cushing secundario a enfermedad adrenocortical nodular pigmentada, que característicamente cursa sin hiperpigmentación tras la adrenalectomía. Se manifiesta generalmente en las primeras dos décadas de la vida de forma atípica, con osteoporosis y estatura baja que condicionan un hábito asténico. El hipercortisolismo intermitente, la ausencia de masas suprarrenales en las pruebas de imagen y un aumento paradójico de la secreción de glucocorticoides tras la supresión con dexametasona pueden dificultar el diagnóstico definitivo, que puede requerir los estudios histológicos32. Otras manifestaciones endocrinas del CC son los adenomas hipofisarios secretores de hormona del crecimiento, corticotropina y prolactina. Hasta el 60% de los pacientes presentan quistes o múltiples nódulos tiroideos en la ecografía, sobre todo adenomas foliculares, que pueden degenerar hasta en el 10% hacia tumores malignos papilares o foliculares. Un 30-50% de los varones con CC pueden desarrollar tumores testiculares, generalmente en las primeras tres décadas de la vida, dando lugar a masas firmes o pétreas al tacto. En el 75% de los casos son multicéntricos y bilaterales. El más frecuente es el de células de Sertoli calcificantes con células grandes (50%), con un bajo potencial de malignización. Este tumor suele manifestarse en la adolescencia con pubertad precoz o ginecomastia, frecuentemente con una exploración física normal. Suele diagnosticarse ecográficamente con la visualización de microcalcificaciones. Este tumor también puede desarrollarse en el síndrome de Peutz-Jeghers. También se han descrito en el CC tumores testiculares de células de Leydig y de células adrenocorticales residuales.
El schwannoma melanocítico psamomatoso es un tumor raro, presente en el 14% de los pacientes, que indica el diagnóstico de CC. Suele localizarse en el tracto gastrointestinal superior, en las cadenas nerviosas simpáticas paravertebrales y, ocasionalmente, en la piel. Son tumores encapsulados y se componen de células de Schwann fusiformes y elongadas con potencial melanogénico. La mayoría de estos tumores son benignos, pero hasta el 10% puede ser maligno, con alta frecuencia de metástasis y pronóstico fatal. Otros tumores asociados al CC son el hepatoma fibrolamelar, el leiomioma mixoide y quistes y carcinoma ováricos.
Sobre la patogenia del CC, se ha especulado que tanto los tumores melanocíticos como los mixomatosos derivarían de restos anormales de células del neurectodermo y del mesodermo, respectivamente, estimulados por concentraciones elevadas de factores de crecimiento similares a la insulina.
El CC es una enfermedad genéticamente heterogénea, de penetrancia variable y expresión incompleta, de la que se ha descrito tanto un patrón de herencia autosómica dominante (50%) como ligado al cromosoma X. En la actualidad, se diferencian dos variedades de CC en función del locus implicado: a) el CC tipo 1, que se produce una inactivación del gen PRKAR1A, en el locus 17q22-24, y se halla hasta en el 65% de los pacientes con CC y el 50% de sus familiares32 –hasta ahora no se han detectado diferencias fenotípicas en los pacientes con esta mutación, aunque sí se ha observado un 80% de familiares portadores de la mutación cuando la forma de presentación del CC es con un síndrome de Cushing33–, y b) el CC tipo 2, en el que el locus alterado se halla en el cromosoma 2p16 pero se desconoce el gen que lo origina5. Otro grupo de pacientes no presenta alteraciones en ninguno de esos dos locus34. Se han identificado algunas variantes fenotípicas del CC que incluyen contracturas congénitas en las que se han detectado mutaciones del gen MYH8, que codifica la miosina perinatal y podría estar implicado en la tumorogénesis de los mixomas cardíacos.
El diagnóstico de CC se puede establecer en estas tres situaciones: a) presencia de dos de las características principales confirmadas por histología, análisis bioquímicos o pruebas de imagen; b) presencia de una de las manifestaciones e historia familiar de CC, y c) paciente portador de una mutación inactivadora del gen PRKAR1A. Hasta en un 50% de los casos de CC la pigmentación cutánea es la primera manifestación. Se recomienda realizar ecocardiografías anuales desde los 5 años de edad a los pacientes, los familiares que tengan por lo menos un criterio diagnóstico del CC aunque estén asintomáticos y los portadores de la mutación del gen PKRAR1A. Del mismo modo, se recomienda la exploración clínica periódica de los testículos y ecografías ováricas regulares. En caso de sospecha de enfermedad adrenal nodular pigmentaria primaria, se recomienda la determinación del cortisol en orina de 24 h y el test de supresión con dexametasona.
En el síndrome NAME, las lesiones pigmentadas aparecen en las primeras semanas de vida, y después se desarrollan los neurofibromas mixoides subcutáneos y, ya en la infancia, los mixomas auriculares. Estos pacientes suelen presentar un fenotipo facial característico, con lentigos faciales e hipertelorismo. Las lesiones pigmentadas más comunes son las efélides, que acostumbran aparecer en los meses de verano, la mayoría de menos de 1 cm y color marrón claro, agrupadas sobre todo en la cara y el cuello y ocasionalmente en el tronco superior y la superficie extensora de los brazos. Entre estas áreas de efélides pueden hallarse máculas similares a manchas de tinta. Las máculas pigmentadas faciales pueden desaparecer con el tiempo35.
Otros síndromes cardiocutáneos. El síndrome de Danoff combina lentigos, mixoma auricular, displasia micronodular corticosuprarrenal y tumores de células fusiformes. El síndrome de preexcitación cardíaca es un síndrome familiar en el que se combinan lentigos cutáneos generalizados con preexcitación cardíaca que da lugar a arritmias y muerte súbita.
LENTIGINOSIS CIRCUNSCRITAS CON TRASTORNOS SISTÉMICOSSíndrome de Peutz-Jeghers (SPJ) o lentiginosis periorificialLas principales características de este síndrome hereditario son lesiones pigmentadas mucocutáneas, poliposis hamartomatosa del tracto gastrointestinal y mayor riesgo de adenocarcinomas en edades tempranas. Su incidencia se cifra36 en 1/30.000-100.000 y puede afectar a ambos sexos y cualquier raza. Se transmite con un patrón de herencia autosómica dominante, y se detectan familiares afectos en el 60% de los casos. Hasta el 80% de los pacientes con SPJ presentan mutaciones en el locus 19p13.3 del gen LKB1/STK11, un gen supresor tumoral de elevada penetrancia y mediador de la muerte celular dependiente de p53 que codifica la serina treonincinasa 1137. El SPJ puede presentarse clínicamente de forma muy variable: desde portadores asintomáticos a enfermos o familiares monosintomáticos o polisintomáticos.
Las lesiones pigmentadas mucocutáneas son el primer signo y la característica más constante del SPJ. Suelen aparecer en los primeros meses o años de vida, aunque también pueden aparecer en la edad adulta. Son máculas marrón oscuro o azules, redondas, ovales o irregulares, de 1-5 mm de diámetro. En ocasiones son de mayor tamaño y semejan máculas café con leche. La localización más frecuente y considerada patognomónica es la mucosa oral, típicamente los labios, en especial el inferior (fig. 8). También pueden observarse en las encías, el paladar duro y la lengua. Otras localizaciones frecuentes de estas máculas son el centro de la cara (peribucales y perinasales), las superficies de las manos y los pies, los
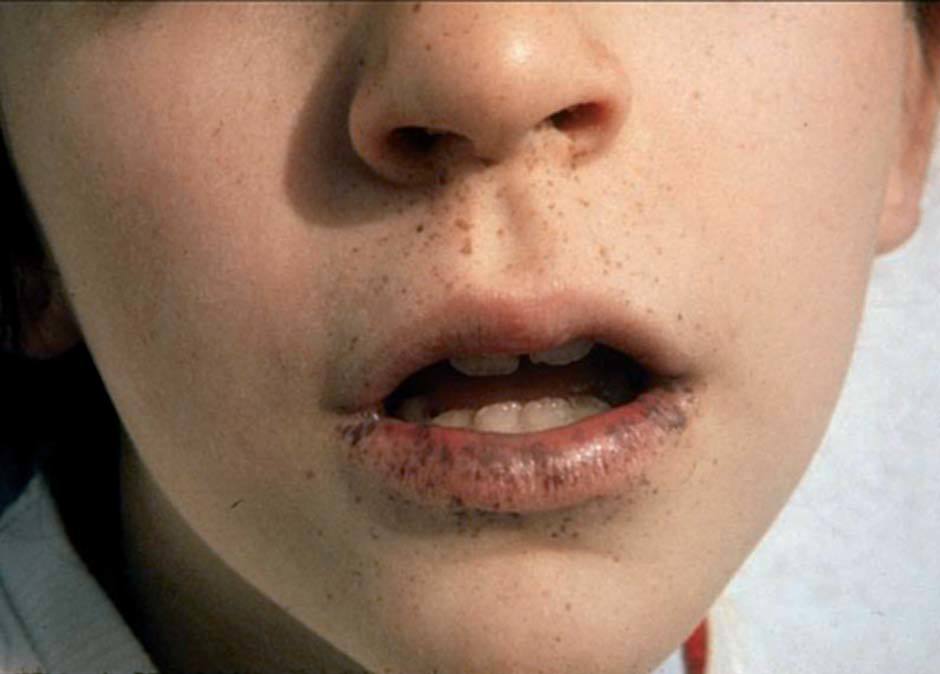
Figura 8. Síndrome de Peutz-Jeghers: lentigos labiales (imagen cedida por el Dr. Bordas, Servei de Dermatologia, Hospital de Bellvitge, L’Hospitalet de Llobregat, Barcelona). ojos, el ombligo y el ano. Se ha descrito puntualmente la aparición de máculas pigmentadas típicas del SPJ en áreas previamente afectadas de psoriasis38. También puede aparecer pigmentación ungueal de manera difusa o en bandas longitudinales. Con excepción de las máculas de la mucosa oral, las demás lesiones pigmentadas pueden desaparecer a partir de la pubertad. Histológicamente, las máculas del SPJ no son realmente lentigos, ya que presentan un aumento en la cantidad de melanina en la capa basal de la epidermis, tanto en los melanocitos como en los queratinocitos, sin cambios en el número de melanocitos. Sin embargo, las lesiones acrales pueden tener un aumento en el número de melanocitos con procesos dendríticos largos llenos de melanosomas. Otras lesiones cutáneas que se pueden hallar en el SPJ son múltiples nevos melanocíticos compuestos, nevos azules, nevos de Spitz y dedos en palillo de tambor.
La poliposis gastrointestinal es otro componente del síndrome y puede desarrollarse en cualquier punto del tracto gastrointestinal, sobre todo en el yeyuno y el íleon. Puede ser la primera manifestación del SPJ, generalmente a partir de los 10-30 años, en forma de ataques de dolor abdominal recurrente secundario a obstrucción intestinal, hemorragias digestivas, diarreas o estreñimiento. Aunque la mayoría de estos pólipos son hamartomas benignos, hay un riesgo de transformación maligna de un 2-3%.
El riesgo de contraer una neoplasia maligna en algún momento de la vida que tienen los pacientes con SPJ es > 90%34, con una media de edad al diagnóstico de 42,9 ± 10,2 años. Los tumores gastrointestinales son los más frecuentes y pueden desarrollarse a partir de pólipos hamartomatosos o de la mucosa normal, en general con mal pronóstico. Los tumores malignos mamarios y genitales son los segundos en frecuencia. El tumor de ovario más frecuente es el de células de la teca granulosa y otros más raros son tumores epiteliales mucinosos y decélulas de Sertoli. Éste, como se ha visto en el CC, también puede hallarse en varones. Otras neoplasias malignas asociadas al SPJ son los tumores de páncreas, tiroides y cuello, el mieloma múltiple y el adenocarcinoma bronquioalveolar. También se han descrito tumores benignos de los tractos respiratorio y urinario.
El diagnóstico de SPJ se realiza con dos de los tres criterios siguientes: a) máculas hiperpigmentadas labiales; b) historia familiar de SPJ, y c) poliposis de intestino delgado. En todo paciente con SPJ se recomienda criba anual de neoplasia intestinal a partir de los 20 años y practicar cada 2 años exploraciones endoscópicas esofagogastroduodenales y pancolónicas, ecografía abdominal y estudios baritados del intestino delgado. Asimismo, se recomiendan exploraciones mamarias y pelvianas a partir de los 25 años para descartar neoplasias mamarias y genitales.
El diagnóstico diferencial del SPJ debe establecerse con la pigmentación mucocutánea melanótica aislada y el síndrome de Bandler. La primera presenta el mismo patrón de pigmentación y el riesgo aumentado de cáncer de mama y genital, pero no incluye poliposis intestinal. El síndrome de Bandler combina el patrón de lentiginosis del SPJ con hemangiomatosis intestinal, que puede dar lugar a graves hemorragias recurrentes.
Síndrome de Bannayan-Riley-Ruvalcaba (BRR)El término BRR abarca tres síndromes de herencia autosómica dominante descritos por separado con características clínicas similares: los síndromes de Riley-Smith, de Bannayan-Zonana y de Ruvalcaba-Myhre-Smith. Se trata de una hamartomatosis que combina la tríada clásica de lentigos genitales, poliposis intestinal y macrocefalia.
Los lentigos genitales aparecen en el pene o la vulva en el nacimiento o entre la primera infancia y la pubertad. También son característicos del BRR los hamartomas vasculares y/o lipomatosos, de rápido crecimiento y comportamiento local agresivo, que se manifiestan como inflamaciones subcutáneas blandas y dolorosas en el tronco y las extremidades. Otras lesiones cutáneas típicas del BRR son pápulas faciales múltiples, en ocasiones queratósicas o papilomatosas, con características histológicas de siringoma o triquilemoma, angioqueratomas y linfangioqueratomas, pápulas queratósicas o papilomas acrales y orales, lesiones faciales similares a la acantosis nigricans y acrocordones múltiples en los pliegues.
La poliposis intestinal se halla en un 35-45% de los casos, con más frecuencia en colon y recto. Los síntomas son frecuentes: diarrea masiva, dolor abdominal, sangrado rectal, anemia crónica, intususcepción y obstrucción intestinal. Otras localizaciones de hamartomas y tumores son las mamas, el tiroides y el sistema nervioso central.
Junto con la macrocefalia, los recién nacidos con síndrome de BRR presentan mayor longitud y peso para la edad gestacional. Otras alteraciones que se han asociado al BRR son dismorfia facial, trastornos oculares, alteraciones osteoarticulares, trastornos del sistema nervioso central y del periférico y retraso del desarrollo motor.
Se han definido unos criterios mínimos para el diagnóstico de BRR: a) macrocefalia; b) tomografía computarizada cerebral normal, y c) como mínimo dos de las siguientes características: familiar de primer grado afecto de BRR, máculas pigmentadas peneanas, pólipos intestinales hamartomatosos, lipomas subcutáneos, miopatía por depósito de lípidos, líneas de Schwalbe y nervios corneales prominentes.
Un 60% de los pacientes con BRR presentan deleciones en la región 10q23.2-q24.1 del gen supresor tumoral PTEN, que codifica una proteína reguladora del desarrollo neurendocrino, y cuya pérdida de función da lugar al desarrollo de hamartomas de las tres capas germinales. Estas mismas mutaciones se han detectado hasta en el 85% de casos de síndrome de Cowden36. Dadas las similitudes clínicas y genéticas entre ambos cuadros, algunos autores consideran que serían diferentes expresiones fenotípicas de un único proceso que se podría denominar síndrome hamartomatoso tumoral PTEN39. Aunque las mutaciones del gen PTEN se han correlacionado con un mayor riesgo de tumores de mama y con la aparición de lipomas, el BRR no conlleva un riesgo aumentado de cáncer. Sin embargo, se recomiendan cribas periódicas de neoplasias malignas y la determinación desde la primera infancia de sangre oculta en heces y hemograma para detectar precozmente posibles hamartomas intestinales en los pacientes con BRR portadores de mutaciones del gen PTEN.
Síndrome de Cronkhite-CanadaEs un proceso adquirido de origen desconocido que cursa con hiperpigmentación cutánea, alopecia y poliposis gastrointestinal.
La hiperpigmentación cutánea se distribuye de manera difusa en las palmas y la superficie volar de los dedos y en forma de máculas en el dorso de las manos. De forma excepcional puede observarse una hiperpigmentación generalizada o lesiones pigmentadas en las mucosas.
La poliposis acostumbra manifestarse a partir de los 50 años con diarrea, dolor abdominal, pérdida de peso y mala absorción grave secundaria a una enteropatía con pérdida de proteínas. A los pocos meses del inicio de los síntomas digestivos, puede aparecer una alopecia parcheada que evoluciona hacia una alopecia total. A la vez, puede aparecer una distrofia ungueal característica, con formación de una uña ventral que excepcionalmente precede en meses o años a los síntomas gastrointestinales. Debe tenerse en cuenta que los pólipos gastrointestinales pueden no detectarse radiológicamente aunque sean sintomáticos.
Lentiginosis neurodisráfica centrofacialEs un síndrome neurocutáneo poco frecuente, transmitido probablemente con un patrón autosómico dominante, que cursa con la tríada de lentigos centrofaciales, trastornos neuropsiquiátricos y malformaciones disráficas (tabla III).

TABLA III. Manifestaciones de la lentiginosis neurodisráfica centrofacial
Los lentigos aparecen progresivamente entre el primer año y los 8-10 años de vida, alcanzando un número variable de 30 a 100 lesiones, para presentar una lenta regresión después. Son pequeñas máculas marrón negruzco que dibujan una banda horizontal en alas de mariposa en el puente de la nariz y en las mejillas. También pueden aparecer en menor número en la frente, los párpados, los labios y la región perioral, respetando las mucosas. El retraso mental se inicia en la edad escolar, pero las alteraciones psiquiátricas suelen desarrollarse más tarde. Las alteraciones esqueléticas y disráficas suelen diagnosticarse en el nacimiento.
Junto con la lentiginosis centrofacial clásica, Greither describió una lentiginosis centrofacial con hipohidrosis, queratodermia palmoplantar, hipodoncia e hiperqueratosis folicular en la línea de implantación del cabello en el cuello.
Lentiginosis con disección arterialEste síndrome fue descrito en dos familias con un posible patrón de herencia autosómica recesiva. Los lentigos generalmente aparecen en la infancia, aumentan en número hasta la edad adulta y luego se atenúan o desaparecen. Suelen distribuirse de manera generalizada respetando las mucosas y predominan en el tronco y las extremidades, sobre todo las inferiores, incluidos palmas, plantas y dedos. Los pacientes afectos tienen predisposición a disecciones de la aorta, la arteria renal y la arteria carótida interna extracraneal, probablemente secundaria a necrosis quística de la media arterial, la cual también tiene origen en la cresta neural.
Síndrome de TayEs un proceso autosómico recesivo, descrito en dos hermanas de Malasia, que se caracteriza por la combinación de lentigos, vitíligo, máculas café con leche, canas prematuras, retraso del crecimiento y mental, cirrosis, aminoaciduria, hiperesplenismo y múltiples alteraciones esqueléticas.
Síndrome FACESEs un trastorno hereditario que se caracteriza por anomalías faciales, anorexia, caquexia, anomalías oculares (eyes) y cutáneas (skin). Los trastornos pigmentarios cutáneos consisten en numerosos lentigos y máculas café con leche.
Facomatosis pigmentovascularSe caracteriza por la combinación de hamartomas melanocíticos y vasculares. Los lentigos son una de las múltiples lesiones pigmentadas que pueden aparecer distribuidas de manera segmentaria. Pueden asociarse a trastornos esqueléticos y del sistema nervioso. También pueden observarse nevus flammeus, nevus spilus y nevo azul, con o sin nevo anémico.
Otras lentiginosisSe han descrito casos aislados de lentiginosis extensas junto con nistagmo y estrabismo, sordera congénita, alteraciones electrocardiográficas, miocardiopatía obstructiva hipertrófica, mixomas auriculares, mixomas cutáneos y aneurisma de la arteria cerebral. Recientemente Ho et al40 han descrito una nueva entidad en la que se conjugaban múltiples lentigos con pigmentación flexural. Por otro lado, durante la instauración de la enfermedad de Addison puede observarse un aumento en el número y en la pigmentación de los lentigos. Otros trastornos que pueden acompañarse con lentigos son la displasia inmunoósea de Schmike, el síndrome de Mul-vihill-Smith, el síndrome de McCune-Albright, las neurofibromatosis I y II y el síndrome MEN2B.
DIAGNÓSTICO DIFERENCIALAnte la sospecha clínica de una lentiginosis, debe realizarse una exploración física completa para determinar si es focal o generalizada y detectar precozmente posibles alteraciones sistémicas concomitantes, sobre todo cardiovasculares y de otras estructuras derivadas de la cresta neural. Inicialmente deben diferenciarse las lentiginosis múltiples de las lentiginosis circunscritas (tabla I). De las lentiginosis múltiples, hay que distinguir fundamentalmente la lentiginosis generalizada, sin trastornos sistémicos, de otras lentiginosis con manifestaciones extracutáneas, como el LEOPARD y el CC. Las dos últimas se diferencian sobre todo porqueel CC cursa con lentigos en las mucosas y tumores mixoides mucocutáneos y no presenta rasgos dismórficos asociados (tabla IV). En el LEOPARD los lentigos faciales son escasos y aparecen las características del acrónimo. El CC debe diferenciarse a su vez del SPJ, una lentiginosis circunscrita con la que comparte la pigmentación mucocutánea, característicamente en el borde del bermellón, y la posibilidad de desarrollar el tumor testicular calcificante de células de Sertoli. Otros diagnósticos diferenciales que pueden plantearse con el CC son la lentiginosis centrofacial, la neurofibromatosis tipo I y el MEN. Respecto a las lentiginosis circunscritas, también deberemos diferenciar las que cursan sólo con pigmentación cutánea de las que cursan además con trastornos sistémicos (tabla I). De las primeras, la LUP debe diferenciarse del nevus spilus, el nevus lentiginoso moteado segmentario y la neurofibromatosis segmentaria. En ocasiones pueden ser útiles la exploración mediante luz de Wood o una biopsia cutánea para comprobar la aparición de lentigos en la piel normal, característica diferencial de la LUP. Si las lesiones de LUP asientan en la cara, el diagnóstico diferencial se establece con el nevo de Ota y otras lentiginosis que cursan con lentigos faciales (tabla V). La lentiginosis centrofacial descrita en la raza negra, a diferencia de la lentiginosis centrofacial clásica, no se combina con otras anomalías extracutáneas y pueden aparecer en las palmas, las plantas y los glúteos lentigos que no desaparecen con el tiempo. Ante pigmentación mucosa y ungueal en una lentiginosis, se debe tener en cuenta los diagnósticos de síndrome de Laugier-Hunziker, el SPJ y el síndrome de Cronkhite-Ca-nada. El síndrome de Laugier-Hunziker es un proceso exclusivamente mucocutáneo sin trastornos sistémicos relacionados. En el SPJ las lesiones pigmentadas ungueales son poco frecuentes y los datos clínicos que deben hacer pensar en él son sobre todo la distribución periorificial de los lentigos y la poliposis concomitante. Estas lentiginosis deben diferenciarse de otros trastornos pigmentarios de la mucosa oral, labial y ungueal como la enfermedad de Addison, en la que la pigmentación es más difusa y puede aparecer en cualquier localización, en pacientes infectados por el VIH y tras la administración de quimioterápicos.

TABLA IV. Diferencias entre el síndrome LEOPARD, el complejo de Carney y el síndrome de Peutz-Jeghers

TABLA V. Lentiginosis con lentigos faciales
CONCLUSIONESLas lentiginosis son un grupo heterogéneo de entidades que ocasionalmente pueden conllevar importantes alteraciones extracutáneas que pueden comprometer la vida de los afectados. Pueden ser un marcador clínico importante, por lo que reconocerlas requiere una minuciosa evaluación del dermatólogo. Dado que algunas de las manifestaciones sistémicas de las lentiginosis son progresivas, se recomienda mantener controles periódicos durante los primeros años de vida con el fin de detectar precozmente anomalías que pueden ser graves (especialmente cardiovasculares o neoplásicas). También será fundamental explorar a los familiares directos y estudiar las mutaciones genéticas causales, que pueden ser útiles para el diagnóstico preciso de algunas formas de lentiginosis. Debe realizarse un consejo genético en los casos en que se establezca un diagnóstico definitivo que implique el potencial desarrollo de complicaciones extracutáneas graves e importantes repercusiones en futuras generaciones.
Correspondencia: Dra. J.M. Sánchez Schmidt. Servei de Dermatologia. Hospital del Mar. Pg. Marítim, 25-29. 08003 Barcelona. España. Correo electrónico: JSanchezS@imas.imim.es