






Una gran parte de la población general (incluyendo numerosos médicos, algunos de ellos dermatólogos) ha subestimado y subestiman todavía el impacto y las consecuencias de las enfermedades cutáneas, olvidando que la piel es un órgano fundamental y como tal desempeña un papel fisiológico tan importante para la vida como el de otros órganos. Muchos se muestran escépticos en relación a la existencia de verdaderas «urgencias dermatológicas», concepto parcialmente desvirtuado en la actualidad en parte porque muchos pacientes son incorrectamente derivados a dermatología de forma «urgente»1.
«Potencialmente fatales» podrían resultar numerosas enfermedades cutáneas de muy variada etiología y características, bien sea porque puedan determinar un compromiso vital inminente o porque condicionen complicaciones tardías relevantes. Es por ello de suma importancia que como clínicos seamos capaces de reconocer y diagnosticar rápidamente estas situaciones, puesto que de ello dependerá el pronóstico del paciente.
Debido al amplio y variado abanico de enfermedades cutáneas que podrían encuadrarse en el concepto que da título a esta revisión, no pretende estructurarse ésta como una recopilación exhaustiva; simplemente aspira a repasar y actualizar las principales características de algunos procesos dermatológicos que por su posible fatalidad requieren una rápida identificación y tratamiento.
Signos de alarmaEn no pocas ocasiones resulta difícil valorar en la primera consulta la gravedad del cuadro que presenta un paciente, ya que la evolución del mismo puede no ser previsible, los síntomas pueden ser escasos en ese instante y el paciente puede no mostrar al inicio una afectación del estado general que transmita una sensación de urgencia.
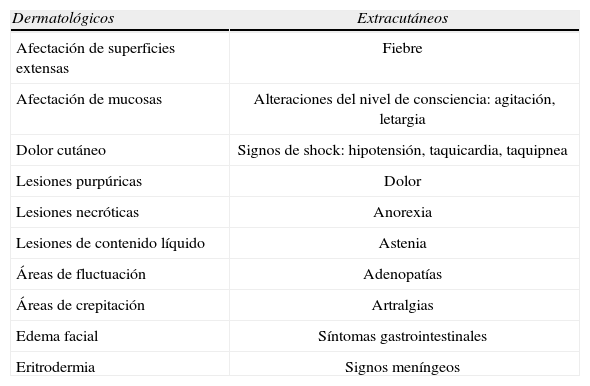
Paralelamente a la propia enfermedad, ciertos condi-cionantes pueden influir en la fatalidad de algunas dermatosis o predisponer a un paciente a desarrollar dermatosis (o complicaciones de éstas) particularmente fatales. Entre aquellos destacan la edad del paciente y sus enfermedades basales (obesidad, HTA, DM, insuficiencia vascular, inmunodepresión). Determinados signos y síntomas cutáneos y sistémicos constituyen importantes datos de alarma que ayudan a intuir una posible evolución fatal (tabla 1). Su presencia hace necesaria la realización de una exploración minuciosa y de las pruebas complementarias apropiadas en cada caso.
Signos y síntomas de alarma en un paciente con lesiones cutáneas
| Dermatológicos | Extracutáneos |
| Afectación de superficies extensas | Fiebre |
| Afectación de mucosas | Alteraciones del nivel de consciencia: agitación, letargia |
| Dolor cutáneo | Signos de shock: hipotensión, taquicardia, taquipnea |
| Lesiones purpúricas | Dolor |
| Lesiones necróticas | Anorexia |
| Lesiones de contenido líquido | Astenia |
| Áreas de fluctuación | Adenopatías |
| Áreas de crepitación | Artralgias |
| Edema facial | Síntomas gastrointestinales |
| Eritrodermia | Signos meníngeos |
Todo clínico debería estar familiarizado con las características de las principales dermatosis que pueden presentar una evolución fatal y rápida y que por ello son subsidiarias de recibir tratamiento hospitalario urgente. Otras muchas que pueden ser fatales a más largo plazo2 (tabla 2) no serán comentadas en las siguientes líneas por limitaciones de espacio.
Clasificación de las dermatosis potencialmente fatales2
| Etiología | Entidades |
| 1. Infecciosa | Síndrome de la piel escaldada estafilocócica |
| Síndrome del shock tóxico | |
| Fascitis necrotizante | |
| Púrpura fulminans | |
| Ectima gangrenoso | |
| Meningococemia diseminada | |
| Rickettsiosis | |
| Micosis profundas | |
| Erupción variceliforme de Kaposi | |
| 2. Farmacológica | Síndrome de Stevens-Johnson/NET |
| DRESS/DIHS | |
| 3. Autoinmune | Pénfigo vulgar |
| Penfigoide ampolloso | |
| Lupus eritematoso sistémico | |
| Dermatomiositis | |
| Esclerodermia | |
| 4. Tumoral | Melanoma |
| Linfomas cutáneos | |
| Angiosarcoma | |
| Carcinoma de células de Merkel | |
| 5. Vascular | Calcifilaxis |
| Enfermedad de Kawasaki | |
| Panarteritis nodosa | |
| Síndrome de Kassabach Merrit | |
| Granulomatosis de Wegener | |
| Síndrome de Churg-Strauss | |
| Síndrome antifosfolípido | |
| 6. Genodermatosis | Epidermolisis ampollosa (juntural de Herlitz, distrófica recesiva de Hallopeau-Siemens) |
| Feto arlequín, bebé collodion | |
| Síndrome de Gardner. Síndrome de Muir-Torre | |
| 7. Miscelánea | Urticaria y anafilaxis |
| Eritrodermia | |
| Insuficiencia cutánea aguda | |
| Psoriasis pustuloso |
DIHS: Drug Induced Hypersensitivity Syndrome; DRESS: Drug reaction/rash with eosinophilia and systemic symptoms; NET: Necrolisis Epidérmica Tóxica.
Concepto. Ambas entidades forman parte de un espectro clínico de infrecuentes pero graves reacciones que cursan con necrosis epidérmica y afectación mucosa. En la actualidad se acepta que presentan rasgos clínicos, etiológicos, demográficos e histológicos que las diferencian del eritema multiforme major3,4, a pesar de que clásicamente se consideró durante mucho tiempo que éste formaba parte del mismo espectro clínico.
Epidemiología. La incidencia del SSJ y de la NET se encuentra entre el 1–7 y el 1–2 casos por millón de habitantes por año, respectivamente5 y es mayor en ciertos grupos de pacientes (ancianos, infectados por el VIH, pacientes con enfermedades sistémicas que habitualmente requieren tratamientos múltiples). El SSJ presenta un 1–3% de mortalidad, mientras que la NET puede alcanzar hasta un 30%5.
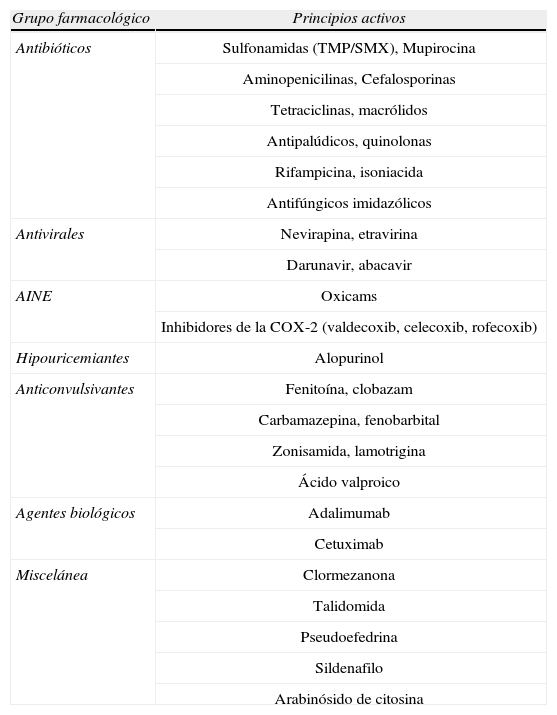
Etiología y etiopatogenia. La mayor parte de los casos de SSJ y NET (aproximadamente 80% y 50–80%, respectivamente) son causados por respuestas idiosincrásicas a fármacos. En la actualidad, más de 220 han sido asociados al desarrollo de estas entidades5,6 (tabla 3). De ellos, el alopurinol es el fármaco implicado con mayor frecuencia7, principalmente cuando se sobrepasan los 200mg diarios. Se han implicado también agentes infecciosos (M. pneumoniae, vacunaciones (sarampión, paperas, rubeola), agentes físicos (radioterapia), enfermedades sistémicas y trasplante de médula ósea5,8–11. Estudios recientes han mostrado una fuerte asociación entre la presencia del alelo HLA‐B*1502 y el desarrollo de SSJ/NET en pacientes asiáticos que reciben tratamiento con carbamazepina12.
Fármacos relacionados con el desarrollo de síndrome de Stevens Johnson (SSJ)/ necrolisis epidérmica tóxica (NET)6
| Grupo farmacológico | Principios activos |
| Antibióticos | Sulfonamidas (TMP/SMX), Mupirocina |
| Aminopenicilinas, Cefalosporinas | |
| Tetraciclinas, macrólidos | |
| Antipalúdicos, quinolonas | |
| Rifampicina, isoniacida | |
| Antifúngicos imidazólicos | |
| Antivirales | Nevirapina, etravirina |
| Darunavir, abacavir | |
| AINE | Oxicams |
| Inhibidores de la COX-2 (valdecoxib, celecoxib, rofecoxib) | |
| Hipouricemiantes | Alopurinol |
| Anticonvulsivantes | Fenitoína, clobazam |
| Carbamazepina, fenobarbital | |
| Zonisamida, lamotrigina | |
| Ácido valproico | |
| Agentes biológicos | Adalimumab |
| Cetuximab | |
| Miscelánea | Clormezanona |
| Talidomida | |
| Pseudoefedrina | |
| Sildenafilo | |
| Arabinósido de citosina |
El desarrollo de SSJ/NET implica una respuesta inmune específica a uno o más fármacos y constituye una forma de hipersensibilidad de tipo retardado13. El suceso fundamental en el SSJ/NET es la apoptosis de los queratinocitos inducida por linfocitos T citotóxicos específicos. La activación del sistema Fas/Fas ligando y la producción de citocinas por los linfocitos T y los macrófagos contribuyen a la patogénesis del SSJ/NET13.
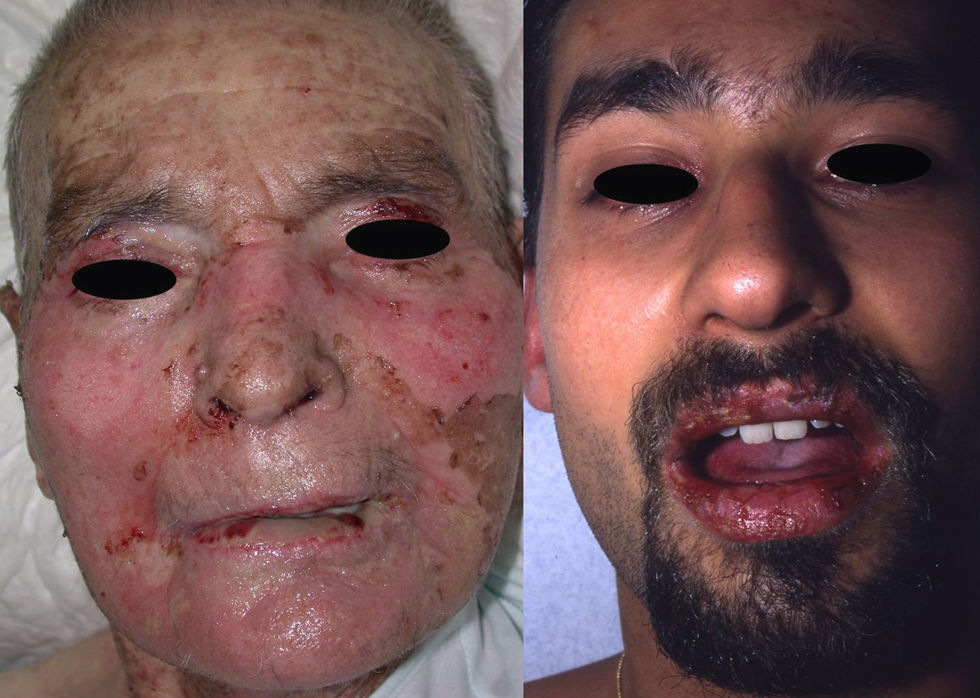
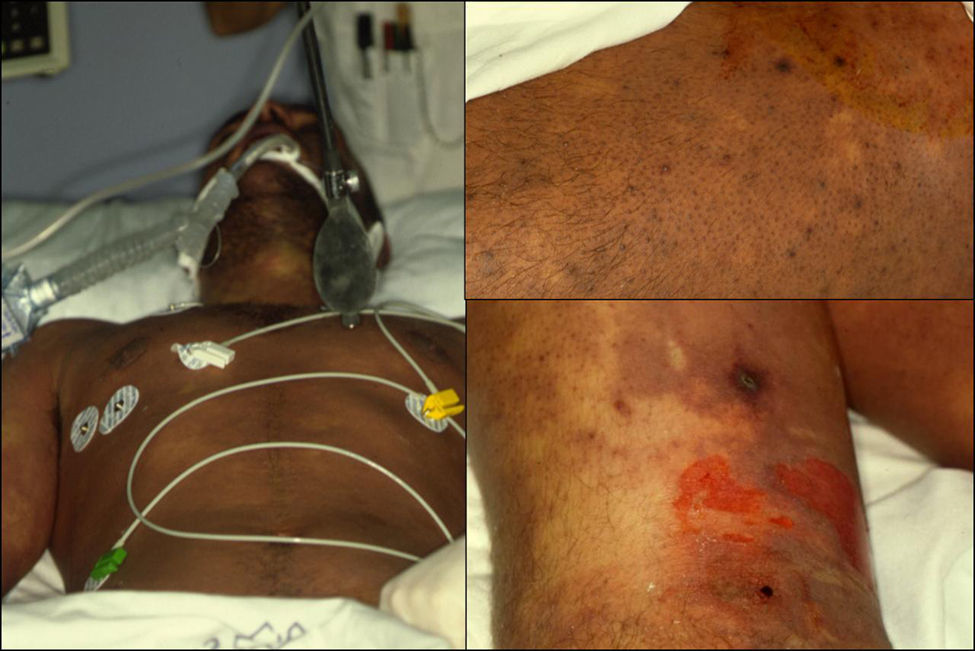
Manifestaciones clínicas. El SJS y la NET se diferencian principalmente en la extensión de las lesiones. El primero se caracteriza por un despegamiento epidérmico inferior al 10% de la superficie corporal total, mientras que la NET se define por un despegamiento que afecta a más del 30% de la misma. Los casos intermedios se denominan «superposición SSJ/NET». Generalmente debutan entre 4–28 días tras la ingesta del fármaco causante, siendo los 2 meses iniciales del tratamiento el periodo de mayor riesgo. Las lesiones cutáneas van precedidas (1–14 días) de una fase prodrómica con síntomas pseudogripales: fiebre, tos, mialgias, artralgias y malestar general. Progresivamente se desarrollan lesiones cutáneas eritemato purpúricas dolorosas, de tamaño y forma irregular (en diana atípica), que se acompañan o siguen de necrosis y desprendimiento epidérmico con ampollas flácidas y signo de Nikolsky positivo4. Se afectan principalmente el tronco y la cara (fig. 1) y en menor medida el cuello y las extremidades. El despegamiento evoluciona a lo largo de 2–15 días y las lesiones van reepitelizando en unos días dejando máculas hipo o hiperpigmentadas6.
 y la semimucosa labial (derecha) de 2 pacientes con necrolisis epidérmica tóxica. Se observan áreas de despegamiento epidérmico.")
Aproximadamente el 90% de los pacientes padece afectación de las mucosas, generalmente en 2 o más territorios, que cursa con áreas eritematosas y erosiones dolorosas. La mucosa oral se afecta casi invariablemente y puede preceder al desarrollo de las lesiones cutáneas. Un 60–85% de los pacientes presenta afectación de la mucosa ocular, que puede determinar importantes secuelas (úlceras corneales, sinequias, xeroftalmia, simblefaron, fotofobia, disfunción de las glándulas de Meibomio e incluso ceguera). Pueden afectarse también las mucosas anogenital, respiratoria y gastrointestinal13.
Diagnóstico. Se fundamenta en la sospecha clínica tras la ingesta de un fármaco sospechoso. El estudio histopatológico muestra la presencia de grados variables de necrosis epidérmica (con afectación de todo el espesor de la epidermis y formación de ampollas subepidérmicas en los casos más graves) y un infiltrado linfocitario cuya densidad se correlaciona con la intensidad del cuadro6. Los estudios de inmunofluorescencia directa son negativos.
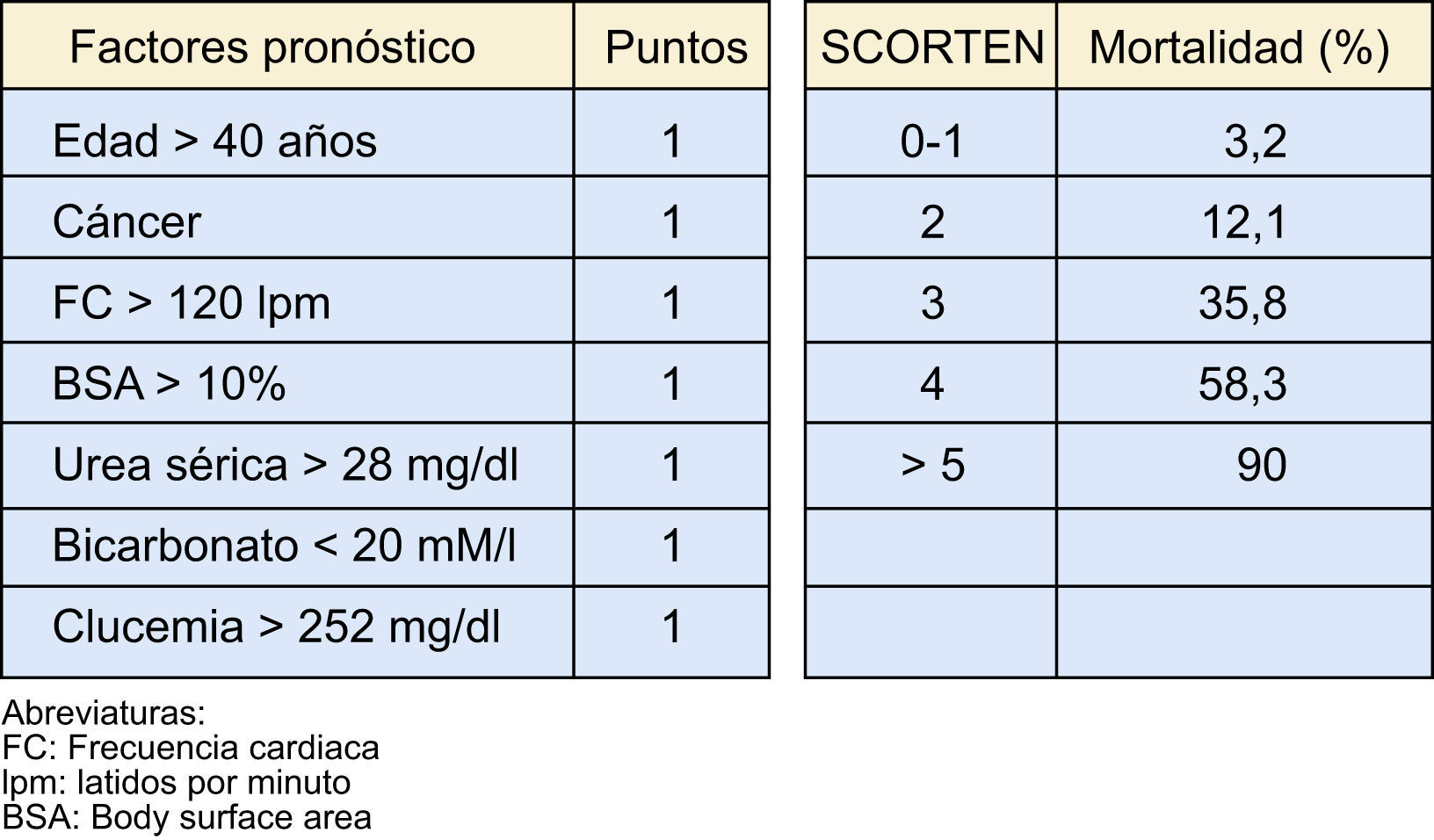
Una herramienta de gran utilidad para estratificar la gravedad del cuadro, especialmente si se realiza de forma precoz (al tercer día), es la escala SCORTEN14 (fig. 2).

Diagnóstico diferencial5. Incluye: síndrome de la piel escaldada estafilocócica (SSSS), eritema multiforme (EM), dermatosis Ig A lineal, pénfigo paraneoplásico, enfermedad de injerto contra huésped aguda, pénfigo y penfigoide inducido por fármacos, exantema fijo medicamentoso generalizado, pustulosis generalizada exantemática aguda15.
Tratamiento. El tratamiento farmacológico de esta entidad sigue resultando controvertido, puesto que no existen evidencias suficientes que permitan destacar el beneficio de un tratamiento sistémico sobre los demás16. La primera y más fundamental medida es la suspensión de cualquier fármaco sospechoso17.
Los pacientes con un SCORTEN superior a 1 deben ser tratados en unidades especializadas18. Se debe realizar un control estricto del balance hidroelectrolítico y de la nutrición del paciente, administrar profilaxis de la enfermedad tromboembólica y analgesia, tomar muestras biológicas repetidas para descartar focos infecciosos (piel, orina, sangre, dispositivos intravasculares) y solicitar una valoración oftalmológica precoz, para evitar el desarrollo de complicaciones oculares difícilmente reversibles.
El tratamiento local consiste en el desbridamiento de las áreas necróticas y protección de las áreas denudadas mediante apósitos biológicos, aloinjertos de piel o apósitos de plata. Éstos deben cambiarse cada 3 días19 y almohadillarse con gasas y vendajes que se cambiarán diariamente.
El tratamiento sistémico4,15 incluye varias opciones:
- •
Corticoides sistémicos: su uso se ha asociado clásicamente al desarrollo de infecciones, retrasos en la cicatrización de las lesiones y aumento de la mortalidad. La mayoría de los expertos desaconsejan su empleo, sin embargo, existen recientes descripciones que sugieren las ventajas de una pauta pulsátil precoz20.
- •
Gammaglobulina intravenosa (IGIV): existen estudios que muestran un incremento de la supervivencia y otros que muestran mínimos o nulos efectos5. Algunos consideran que dada la escasa toxicidad de la IGIV y considerando la gravedad de la situación, el balance riesgo/beneficio favorece el inicio de este tratamiento6.
- •
Plasmaféresis: permite eliminar el factor patogénico del torrente sanguíneo. Los resultados obtenidos, al igual que con otras alternativas, varían de un estudio a otro6.
- •
Tratamiento combinado: algunos autores han observado una reducción de la mortalidad con la administración conjunta de IGIV y corticoides21.
- •
Antibióticos sistémicos: deben prescribirse únicamente ante la presencia de un foco infeccioso y no de forma profiláctica.
- •
Algunos autores recomiendan la administración de factor estimulador de las colonias de granulocitos cuando las cifras de neutrófilos desciendan por debajo de 1000 por mm3,22.
También se han descrito respuestas variables tras la administración de ciclofosfamida, ciclosporina, infliximab y pentoxifilina6.
Síndrome de hipersensibilidad inducido por fármacos (SHIF)Concepto. Es una reacción grave inducida por fármacos que cursa con afectación cutánea y multisistémica. Se denomina también DRESS (drug reaction /rash with eosinophilia and systemic symptoms).
Etiología y etiopatogenia. Los anticonvulsivantes son los fármacos asociados con mayor frecuencia al desarrollo de este cuadro. La dapsona, la salazosulfapiridina, la mexiletina, la minociclina, el metimazol, la cianamida y el alopurinol también pueden causarlo. El virus herpes humano 623, el virus herpes humano 724, el virus de Epstein-Barr25 y el citomegalovirus26 podrían estar implicados en el desarrollo de esta entidad.
Se considera que el SHIF se debe a una reacción inmunológica al fármaco o a alguno de sus metabolitos, aunque este mecanismo no es suficiente para explicar el comportamiento global de la enfermedad.
Manifestaciones clínicas. Se caracteriza por la aparición de fiebre, edema facial y un exantema máculo-papuloso que se inicia entre 2–6 semanas tras la administración del fármaco causante. Pueden aparecer adenopatías, hepatitis, fallo renal y alteraciones hematológicas (linfocitosis atípica, eosinofilia, leucocitosis). Se observa con frecuencia un descenso en los niveles de IgA, IgG e IgM27. Los síntomas pueden mantenerse durante más de 2 semanas tras la suspensión del mismo y no son infrecuentes los rebrotes27,28. Las complicaciones de este cuadro son raras e incluyen encefalitis, hipotiroidismo, miocarditis y diabetes mellitus. Su mortalidad puede llegar a ser del 20% y se correlaciona con el grado de afectación hepática y renal27.
Diagnóstico. Shiohara et al establecieron en el año 2007 los criterios diagnósticos de esta entidad29: 1) exantema maculo-papuloso que aparece más de 3 semanas tras el inicio del fármaco causante; 2) persistencia de los síntomas más allá de 2 semanas tras la suspensión del agente causal; 3) fiebre (>38°C); 4) alteraciones hepáticas (elevación de transaminasas, especialmente GGT); 5) alteraciones leucocitarias (al menos una de las siguientes): leucocitosis (>11×109L−1); linfocitosis atípica (>5%); eosinofilia (>1,5×109L−1); 6) linfadenopatía y 7) reactivación del VVH6. Para el diagnóstico de un SHIF típico es necesaria la presencia de los 7 criterios. Las pruebas epicutáneas y el test de transformación de linfocitos realizados tras la remisión del cuadro suelen mostrar resultados positivos en la mayoría de los pacientes. En el estudio histopatológico suele ser habitual la presencia de infiltrados linfocitarios (perivasculares superficiales y en ocasiones en banda con epidermotropismo), con extravasación de eritrocitos y eosinófilos.
Tratamiento. No existe un tratamiento protocolizado. A pesar de ello, los corticoides constituyen hoy en día la alternativa más empleada (40–60mg de prednisolona, con descenso gradual en 6–8 semanas; pulsos de metilprednisolona iv, 30mg/kg durante 3 días)27. En los casos de SHIF asociado a infección por CMV debe administrarse tratamiento con antivirales (ganciclovir o foscarnet)30.
Dermatosis potencialmente fatales de origen infecciosoSíndrome de la piel escaldada estafilocócica (SSSS) o enfermedad de RitterConcepto. El SSSS es un cuadro clínico desencadenado por la liberación de toxinas exfoliativas producidas por el Staphylococcus aureus.
Epidemiología. Afecta mayoritariamente a niños, con una incidencia de 0,09–0,13 casos por millón de habitantes por año31. Los pacientes inmunodeprimidos y aquéllos con insuficiencia renal poseen un mayor riesgo de padecerlo. Se ha descrito una mortalidad de hasta el 11% en niños y del 40–60% en adultos31,32.
Etiología y etiopatogenia. Está producido por exotoxinas exfoliativas (exfoliatinas A y B), sintetizadas por algunas cepas de S. aureus (fago grupo II, tipos 3a, 3b, 3c, 55, 71) y más raramente por cepas de S. aureus meticilin-resistente, que se unen a la desmogleína 1 y producen un despegamiento intraepidérmico entre los estratos espinoso y granuloso33.
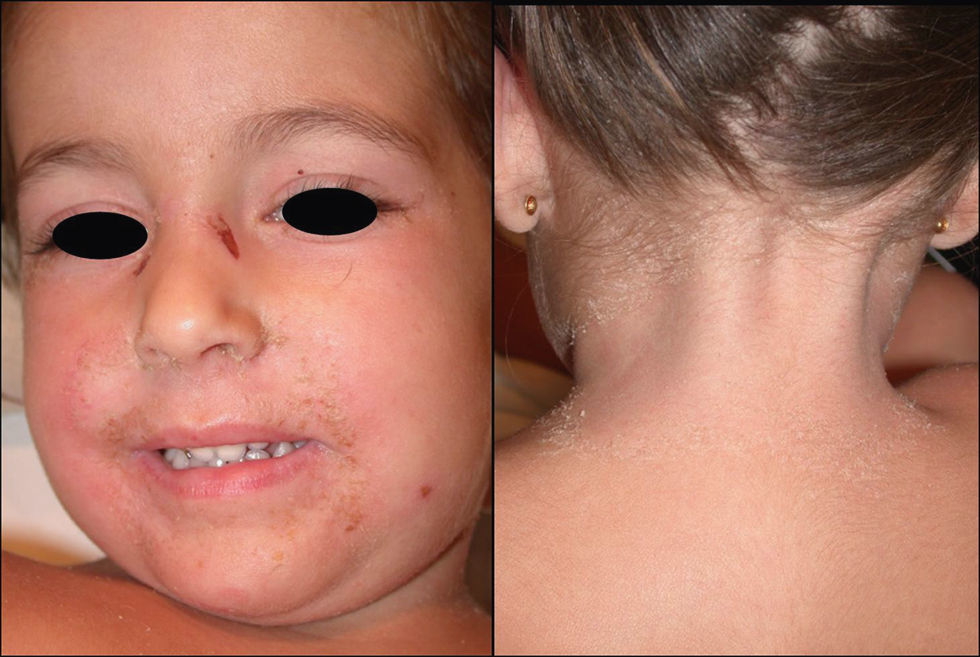
Manifestaciones clínicas. Se presenta de forma abrupta con un eritema de tonalidad anaranjada (fig. 3), difuso y mal delimitado, acompañado de malestar general, hipersensibilidad o dolor cutáneo y fiebre. El cuadro evoluciona produciendo amplias zonas de despegamiento epidérmico con signo de Nikolsky positivo que dejan al descubierto áreas erosivas exudativas. Característicamente respeta las mucosas y se acentúa en las áreas periorificiales y en las flexuras (fig. 4). Suele asociarse a la presencia de un foco infeccioso (conjuntivitis, otitis, faringitis, artritis séptica o piodermitis). Generalmente el cuadro se resuelve en 5–14 días con tratamiento adecuado. Las complicaciones son infrecuentes e incluyen neumonía, celulitis, desequilibrio hidroelectrolítico/térmico y sepsis.


Diagnóstico diferencial. Debe establecerse con el impétigo ampolloso, las quemaduras térmicas y por químicos, la NET, la epidermolisis ampollosa, la escarlatina, el eritema multiforme, la enfermedad de Kawasaki, la eritrodermia ictiosiforme congénita ampollosa y el síndrome de shock tóxico.
Tratamiento. Deben instaurarse medidas generales que incluyan la reposición hidroelectrolítica y el control de la temperatura, la cura de las lesiones cutáneas, la realización de análisis de sangre con marcadores inflamatorios, la recogida de muestras para cultivos (hisopos de las lesiones cutáneas, hisopos nasales, hemocultivos) y la administración de la analgesia necesaria. El midazolam intravenoso puede ser útil si el paciente presenta ansiedad y la gabapentina puede ayudar a controlar el prurito32.
Es necesario también un tratamiento específico con antibióticos sistémicos (bencilpenicilina o macrólidos) y con antibióticos tópicos en los focos infecciosos (conjuntivitis, lesiones cutáneas). En los casos con importante repercusión sistémica puede ser útil administrar plasma fresco congelado (10ml/kg), que contribuye a la neutralización de las toxinas circulantes32.
Síndrome del shock tóxico (SST)Concepto. El SST es una enfermedad aguda, multisistémica y muy grave, ocasionada por cepas de S. aureus y Streptococcus pyogenes (Streptococcus del grupo A) productoras de toxinas.
Epidemiología. El SST estafilocócico menstrual se describió clásicamente en relación al uso de tampones muy absorbentes durante la menstruación. Presenta una incidencia de 0,9–3,4 casos por 100.000 habitantes por año. El SST no menstrual es más prevalente, constituyendo aproximadamente el 62% del total de SST estafilocócicos y presenta una mayor mortalidad que el menstrual34,35.
El SST estreptocócico presenta una incidencia de 1,5–5,2 casos por 100.000 habitantes por año y una mortalidad del 23–80%35.
Etiopatogenia. Ambos SST son secundarios a focos de colonización o infección por cepas productoras de toxinas. El SST estafilocócico no menstrual puede aparecer tras alteración de la barrera cutánea o mucosa, asociado a abscesos o quemaduras y tras intervenciones quirúrgicas; el SST estraptocócico se origina generalmente en focos de fascitis necrotizante, celulitis y miositis. Las toxinas producidas por estas bacterias se comportan como superantígenos, desencadenando una activación extrema de las células T que determina una producción exagerada de citocinas. Las toxinas más comúnmente asociadas al desarrollo del SST son la TSST-1, las enterotoxinas B y C (estafilocócicas) y la exotoxina pirogénica estreptocócica A.
Manifestaciones clínicas. El SST estafilocócico debuta de forma brusca con un cuadro pseudogripal (fiebre, mialgias, cefalea, odinofagia, náuseas, vómitos) seguido de confusión, letargia, agitación e hipotensión. En las primeras 48h suele observarse un exantema maculoso escarlatiniforme centrífugo, que en 1–2 semanas da paso a descamación. Es habitual la presencia de hiperemia conjuntival, faringitis, lengua en fresa y erosiones en la mucosa oral y los labios. El cuadro progresa tan rápidamente que el fallo multiorgánico puede ser evidente en 8–12h35,36. Entre las complicaciones de este cuadro figuran fallo renal, alteraciones de la función hepática, síndrome del distress respiratorio del adulto, cardiomiopatía, encefalopatía y coagulación intravascular diseminada37.
En el SST estreptocócico los síntomas iniciales pueden variar en función del foco infeccioso. Cuando éste asienta en la piel y los tejidos blandos, suele iniciarse con dolor localizado acompañado de eritema y edema, en ocasiones con hematomas y ampollas hemorrágicas35–37. En un 20% de los pacientes pueden observarse síntomas pseudogripales (fiebre, odinofagia, adenopatías, náuseas y vómitos) e hipotensión (fig. 5). Casi el 80% de los pacientes presentan afectación de la función renal37.

Diagnóstico. Se basa en la sospecha y los hallazgos clínicos (tabla 4). La realización de hemocultivos proporciona resultados positivos en más del 60% de los pacientes con SST estreptocócico y en menos del 5% de los pacientes con SST estafilocócico.
Criterios diagnósticos del Síndrome del shock tóxico35,37
| SST estafilocócico | SST estreptocócico |
Generales
| Clínicos:
|
| 6. Laboratorio: resultados negativos en | Laboratorio |
|
|
BUN: Nitrógeno ureico; CPK: Creatinfosfokinasa; FN: Fascitis necrotizante; LCR: Líquido cefalorraquídeo; TAS: Tensión arterial sistólica; SBGA: Streptococcus β-hemolítico del grupo A; SDRA: Síndrome del distress respiratorio del adulto; SNC: Sistema nervioso central; x2: Dos veces por encima del límite superior de la normalidad.
Diagnóstico:
SST estafilocócico: Probable si se cumplen 5 de los 6 hallazgos descritos; definitivo si se cumplen los seis.
SST estreptocócico: Probable si se cumplen todos los criterios clínicos y se aísla SBGA de una localización no estéril; definitivo si se cumplen todos los criterios clínicos y se aísla SBGA de una localización estéril.
Diagnóstico diferencial. Incluye sepsis causadas por otros microorganismos, fiebre manchada de las Montañas Rocosas, fiebre tifoidea, meningococemia, escarlatina, enfermedad de Kawasaki, leptospirosis y SSJ/NET37.
Tratamiento. Es fundamental la instauración rápida de medidas de soporte y de un tratamiento antibiótico específico. Cuando no haya sido posible identificar el agente causal, la pauta debe cubrir Staphylococcus y Streptococcus. Los fármacos de primera línea son la penicilina G, las cefalosporinas de primera generación, la nafcilina, la oxacilina, la clindamicina, la claritromicina y la rifampicina para el S. aureus sensible a meticilina; la clindamicina, el linezolid, la rifampicina, la vancomicina y la teicoplanina para el S. aureus meticilina resistente y la penicilina G, la clindamicina, la vancomicina y la teicoplanina para el S. pyogenes35. El tratamiento debe administrarse durante 10 días, salvo en casos de infecciones profundas como la osteomielitis, que requieren pautas más prolongadas. Estudios recientes han sugerido el posible beneficio de la administración de gammaglobulina intravenosa adyuvante38.
Fascitis necrotizanteConcepto. Es una infección de rápida evolución que afecta a la fascia y al tejido celular subcutáneo39,40.
Epidemiología. Presenta una incidencia de 1–4 casos por millón de habitantes y año y una mortalidad elevada (15–35%). La mayor parte de los casos aparecen en pacientes con procesos subyacentes que empeoran el pronóstico41, entre los que figuran DM, alcoholismo, gota, neoplasias, insuficiencia vascular periférica, insuficiencia renal, infección por el VIH, malnutrición, cirrosis, tratamiento crónico con corticoides, alcoholismo y edad avanzada37,42.
Etiología y etiopatogenia. En función de los microorganismos causantes se distinguen 3 tipos de fascitis necrotizante42: la de tipo II es monomicrobiana, causada por S. pyogenes solo o en asociación con Staphylococcus; la de tipo III está causada por Aeromonas spp. y Vibrio spp.; la de tipo I es polimicrobiana y representa la mayor parte de los casos37,42. Entre los microorganismos causantes se encuentran Staphylococcus, enterococos, Bacteroides, Clostridium spp., Peptostreptococcus, P. mirabilis, P. vulgaris, P. aeruginosa, Klebsiella pneumoniae, E. coli, Morganella morganii, Serratia marcencens, Acinetobacter spp., Enterobacter cloacae, Salmonella y H. influenzae42.
La mayoría de los pacientes presentan condiciones predisponentes (procedimientos quirúrgicos, lesiones cutáneas, abscesos o traumatismos).
La invasión del tejido blando por estos microorganismos determina alteraciones en la oxigenación del mismo que alteran el funcionamiento de los leucocitos polimorfonucleares y facilitan la proliferación bacteriana. La hipoxia y la liberación de productos bacterianos que actúan como superantígenos conducen a una progresiva necrosis tisular y a la aparición de numerosos síntomas sistémicos37.
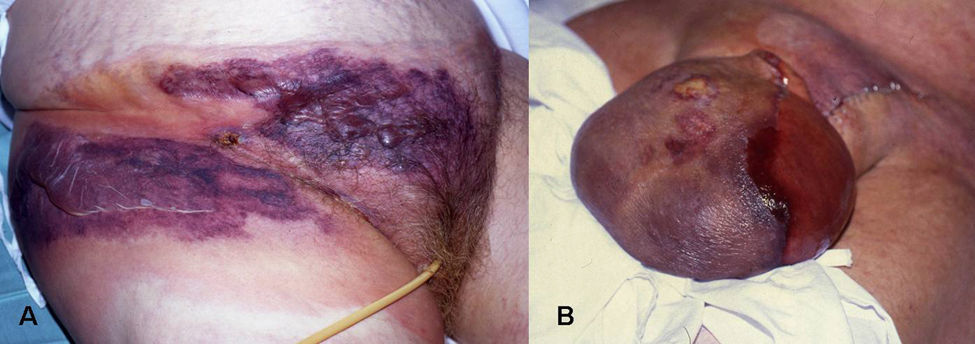
Manifestaciones clínicas. Puede presentarse en cualquier localización, aunque con mayor frecuencia afecta extremidades, tronco (fig. 6A) y periné (gangrena de Fournier), (fig. 6B). Los síntomas iniciales pueden confundirse con una celulitis (eritema, aumento de la temperatura local, edema y dolor), lo que a veces determina un retraso diagnóstico importante y empeora el pronóstico del paciente. Se debe sospechar el cuadro ante la presencia de: dolor muy intenso (característicamente desmesurado en relación a la apariencia clínica de las lesiones), ampollas hemorrágicas, equimosis, necrosis, crepitación, fluctuación, edema indurado que sobrepasa el margen visible del eritema, alteraciones locales de la sensibilidad, progresión rápida y síntomas sistémicos. Éstos incluyen fiebre, alteraciones de la consciencia, taquicardia, taquipnea, fallo multiorgánico y coagulación intravascular diseminada40.
 Fascitis necrotizante. Placa en abdomen e ingle, con amplias zonas equimóticas y ampollas flácidas serohemorrágicas. B) Gangrena de Fournier.")
Diagnóstico. El reconocimiento precoz de una fascitis necrotizante puede ser difícil incluso para clínicos experimentados. Ante la sospecha clínica, la realización de una exploración quirúrgica de la zona afectada es de suma importancia, puesto que permite valorar el aspecto del tejido subcutáneo y la fascia y obtener material para cultivo. La radiografía de las partes blandas puede mostrar gas y la tomografía computarizada y la resonancia magnética nuclear pueden revelar la presencia de edema en los tejidos blandos. El análisis de sangre suele mostrar leucocitosis y elevación de los reactantes de fase aguda.
Tratamiento. La combinación de un tratamiento quirúrgico con una pauta adecuada de antibióticos sistémicos es el tratamiento de elección. Se requiere un rápido y amplio desbridamiento de los tejidos necróticos siendo necesario en algunas ocasiones recurrir a la amputación. La pauta antibiótica ha de iniciarse de forma inmediata y debe cubrir gérmenes Gram positivos, Gram negativos y anaerobios. Una de las pautas más empleadas incluye penicilina G/ampicilina-sulbactam (aztreonam o vancomicina en caso de alergia)+clindamicina+ciprofloxacino/aminoglucósido. En caso de shock pueden ser útil la administración de IGIV y anticuerpos monoclonales anti-TNFα37. Deben pautarse las medidas de soporte necesarias para el control del equilibrio hidroelectrolítico, la temperatura y la nutrición del paciente.
Púrpura fulminans (PF)Concepto. Es un síndrome agudo de etiología variada y elevada mortalidad que cursa con afectación del estado general y de la coagulación, y con lesiones cutáneas purpúricas.
Etiología y etiopatogenia. La PF de origen infeccioso es la forma más común. Se ha asociado a infecciones por N. meningitidis, S. pneumoniae, Klebsiella, H. influenzae, H. aegyptius, S. aureus, S. β-hemolítico del grupo A, P. aeruginosa, Rickettsia spp., C. albicans, E. coli43.
La PF postinfecciosa generalmente se presenta entre 7–10 días después de infecciones víricas (varicela, HHV644) o estreptocócicas.
Otras formas de PF pueden ser debidas a un déficit congénito de proteínas C y S y de antitrombina III, o a un déficit adquirido de proteínas C y S, a la ingesta de fármacos (paracetamol, derivados cumarínicos, propiltiouracilo45 o puedan desarrollarse en el contexto de diversos procesos (síndrome nefrótico, diálisis peritoneal, trasplante de médula ósea).
También existe una PF asociada a enfermedades sistémicas: lupus eritematoso sistémico, panarteritis nodosa, púrpura de Schönlein-Henoch, síndrome antifosfolípido.
El proceso patológico primario de este cuadro es el desarrollo de una coagulación intravascular diseminada. En los casos producidos por N. meningitidis la liberación de endotoxinas activa la cascada de la coagulación y la producción de mediadores inflamatorios. En el desarrollo de la PF postinfecciosa influye de forma determinante un déficit autoinmune adquirido de proteína S43 acompañado de hipofibrinogenemia46.
Manifestaciones clínicas47. Son características las lesiones cutáneas purpúricas palpables y dolorosas, de tamaño variable, rodeadas de un halo eritematoso. Las extremidades inferiores son las zonas más afectadas; la cara y el tronco tienden a presentar menor afectación. Estas lesiones confluyen hasta formar áreas equimóticas en el transcurso de unas horas. El proceso avanza ocasionando úlceras, zonas de isquemia e infartos digitales. Los síntomas cutáneos se acompañan de una marcada afectación del estado general con fiebre, náuseas, vómitos, alteraciones del nivel de consciencia e hipotensión. Los casos infecciosos suelen mostrar mayor afectación hemodinámica.
Diagnóstico. Se basa en las manifestaciones clínicas. Las pruebas de laboratorio muestran hallazgos concordantes con una coagulopatía de consumo: disminución del fibrinógeno, las plaquetas, los factores V y VII, las proteínas C y S y la antitrombina, elevación del dímero D y prolongación de los tiempos de protrombina y de tromboplastina.
Diagnóstico diferencial. El diagnóstico diferencial incluye la púrpura de Schönlein-Henoch, la panarteritis nodosa, la crioglobulinemia y el edema hemorrágico agudo.
Tratamiento47,48. Davis et al47 recopilaron recientemente los principios del tratamiento de la PF. Debe incluir antibióticos cuando la causa del cuadro sea infecciosa (las cefalosporinas de tercera generación son de elección en casos de N. meningitidis) y medidas de soporte intensivas para controlar la hipotensión, el shock y la coagulación intravascular diseminada (corrección de las alteraciones hidroelectrolíticas y del pH, control de las constantes vitales y de la oxigenación). Deben realizarse curas de las lesiones cutáneas, con desbridamiento de las zonas necróticas y fasciotomías y/o amputaciones cuando sea preciso. La administración de heparina, la infusión de plasma fresco congelado, la antitrombina III, la plasmaféresis y los agentes anti-TNF han resultado beneficiosas37,46.
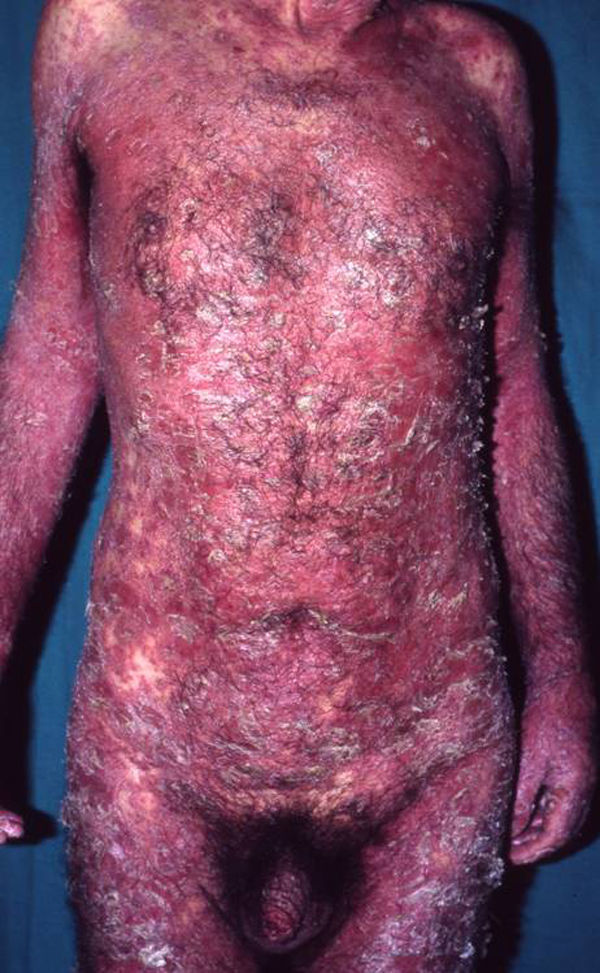
EritrodermiaConcepto. Eritema y descamación en grado variable que afectan a más del 90% de la superficie corporal total. Supone el 1% de todos los ingresos hospitalarios49.
Etiología. Numerosas enfermedades cutáneas pueden manifestarse como una eritrodermia. Las más comunes son la psoriasis, la dermatitis atópica, las toxicodermias y los linfomas cutáneos de células T. Otras causas incluyen pitiriasis rubra pilaris, dermatitis alérgica de contacto, fotodermatosis, enfermedades ampollosas, sarna noruega, liquen plano, enfermedades del tejido conectivo, enfermedad de injerto contra huésped, ictiosis, infecciones, dermatitis seborreica50. Existen también casos idiopáticos y casos paraneoplásicos producidos fundamentalmente por neoplasias hematológicas.
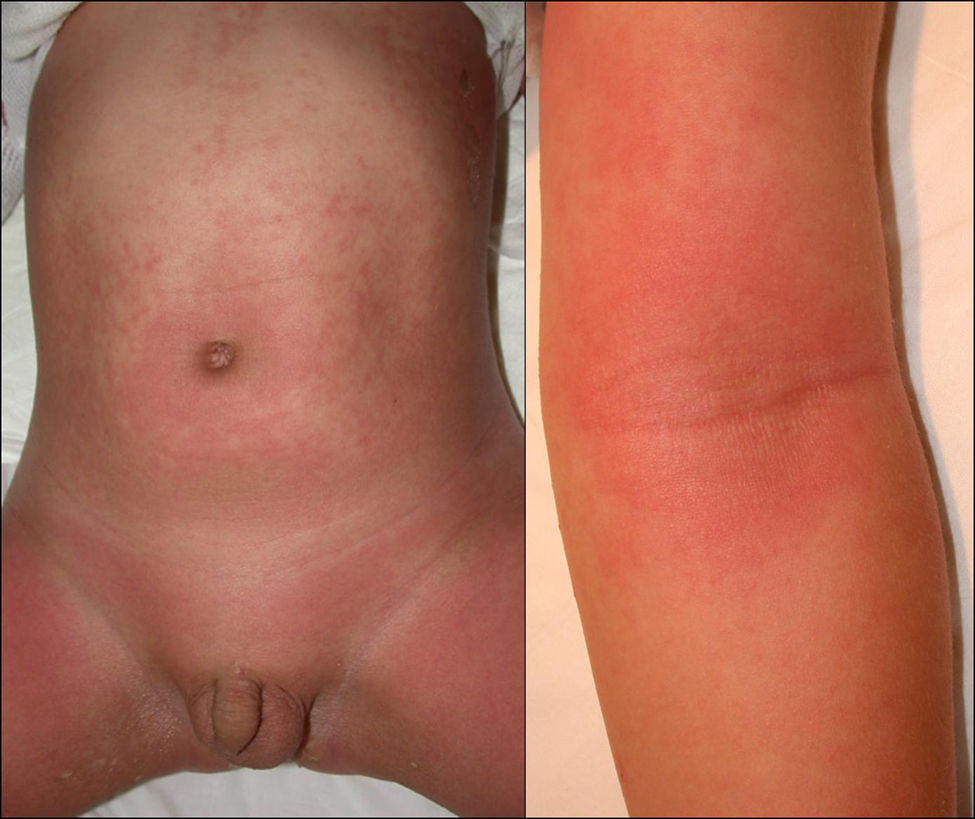
Manifestaciones clínicas. Eritema y descamación generalizados (fig. 7) que pueden acompañarse de prurito, sensación de escozor, fatiga, mal estar general y fiebre. Pueden observarse pérdida de cabello, alteraciones ungueales, ectropion, alteraciones de la pigmentación, linfadenopatías, edema y queratosis seborreicas eruptivas transitorias51. Además pueden presentarse síntomas sistémicos como hipoalbuminemia, pérdida de peso, alteraciones hidroelectrolíticas graves, síndrome del distress respiratorio del adulto, desequilibrio térmico, infecciones e insuficiencia cardíaca de alto gasto52. Su tasa de mortalidad varía entre 18–64%49.

Diagnóstico. Se basa en los hallazgos clínicos y anatomopatológicos. Debe realizarse una historia clínica minuciosa prestando especial atención a la posible existencia de fármacos desencadenantes. Los análisis de sangre, el cultivo de muestras biológicas y las pruebas de imagen pueden resultar de utilidad para orientar la causa.
Tratamiento. El tratamiento etiológico variará en función del proceso causante y debe complementarse siempre con medidas de soporte vital. De forma tópica pueden ser útiles los emolientes y los corticoides.
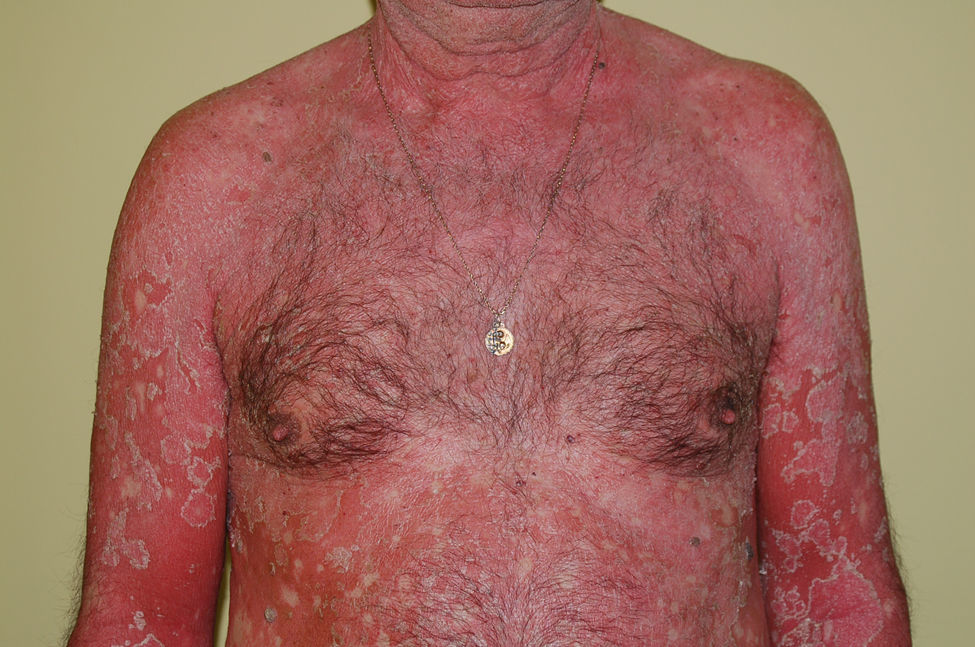
Insuficiencia cutánea agudaConcepto. Situación que resulta de la alteración de las diferentes funciones de la piel (termorreguladora, inmunológica, metabólica, neuroendocrina y de relación con el medio exterior) y que conduce al desarrollo de diversas complicaciones que comprometen la vida del paciente53.
Etiología. Los cuadros que con mayor frecuencia la ocasionan son toxicodermias, enfermedades ampollosas, quemaduras extensas y psoriasis pustuloso. Cualquier dermatosis que se presente como o evolucione hacia una eritrodermia puede conducir a una insuficiencia cutánea aguda (fig. 8). Como consecuencia de la alteración de las funciones de la piel se producen entrada de gérmenes en el organismo, pérdida de agua, electrólitos y proteínas, distermia y alteraciones inmunológicas.

Manifestaciones clínicas. Las manifestaciones sistémicas que forman parte de este síndrome son las que secundariamente corresponden a la alteración de cada una de las funciones de la piel enumeradas anteriormente. Pueden liberarse hormonas de estrés que incrementan el catabolismo y pueden producir resistencia a la insulina4. Los hallazgos cutáneos varían en función de la dermatosis de base.
Tratamiento. El tratamiento etiológico depende de la causa. Es fundamental instaurar las medidas de soporte necesarias para garantizar la nutrición del paciente, su equilibrio térmico e hidroelectrolítico, mantener sus constantes vitales y evitar posibles complicaciones53,54.
Es preciso recordar también que los efectos de diversos tratamientos sistémicos dermatológicos pueden ser potencialmente fatales. Dado que las lesiones cutáneas pueden constituir la primera manifestación de un cuadro de evolución fatal (sea cutáneo primario o sistémico), el dermatólogo desempeña un papel fundamental en el diagnóstico, seguimiento y tratamiento de estos procesos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
- 1.
Es fundamental para el dermatólogo sospechar y saber reconocer las principales dermatosis que pueden comprometer de forma inminente la vida del paciente.
- 2.
Es necesario prestar atención a la existencia de determinados síntomas y signos cutáneos y sistémicos que puedan indicar la presencia de una dermatosis de mayor gravedad.
- 3.
Del amplio abanico de dermatosis potencialmente fatales destacan por su rapidez de evolución las de origen farmacológico e infeccioso.
- 4.
La mayoría de estos cuadros requieren tratamiento en el medio hospitalario con estricto control de las constantes vitales, del equilibrio hidroelectrolítico, la nutrición y la temperatura del paciente.
- 5.
Aproximadamente un 30% de las toxicodermias son cuadros graves, con compromiso vital. En su evolución influyen la edad, el tipo de fármaco implicado y el grado de afectación clínica.
- 6.
El SSSS suele cursar de forma benigna en niños; en adultos, sin embargo, presenta una elevada mortalidad. Al igual que el SST, es un cuadro ocasionado por la liberación de exotoxinas.
- 7.
La fascitis necrotizante puede simular inicialmente una celulitis, por lo que la sospecha clínica en un paciente con antecedentes de riesgo y el inicio rápido de tratamiento médico quirúrgico son imprescindibles para la supervivencia del paciente.
- 8.
Diferentes dermatosis pueden desencadenar una eritrodermia. Las causas más frecuentes son la psoriasis, la dermatitis atópica, los linfomas cutáneos y las toxicodermias.
- 9.
La piel es tan importante para la supervivencia como otros órganos. La alteración de sus funciones básicas conduce a una insuficiencia cutánea aguda, cuyo desenlace puede ser fatal.
- a)
Los pacientes con un SCORTEN superior a 5 presentan una mortalidad superior al 90%.
- b)
Se han descrito casos relacionados con la administración de nevirapina y cotrimoxazol.
- c)
Prácticamente todos los pacientes presentan afectación de la mucosa oral y un 60–85% de ellos pueden presentar afectación ocular.
- d)
Debe iniciarse precozmente una pauta profiláctica de antibióticos sistémicos para evitar complicaciones.
- e)
Es un cuadro muy infrecuente, con una incidencia de 1–2 casos por millón de habitantes por año.
Comentario: El tratamiento de la NET no se encuentra protocolizado. Las últimas recomendaciones, sin embargo, desaconsejan el empleo sistemático de antibióticos sistémicos sin evidencia de un foco infeccioso activo. Pueden emplearse apósitos impregnados de antibióticos o de plata para realizar las curas de las lesiones cutáneas.
2. Es falso en relación con la fascitis necrotizante- a)
Frecuentemente se presenta de forma brusca en pacientes sanos sin antecedentes de interés.
- b)
Los estreptococos son los agentes más frecuentemente implicados en el cuadro.
- c)
Es habitual observar una discordancia entre la severidad del cuadro y sus manifestaciones clínicas.
- d)
Es una de las infecciones cutáneas con mayor morbimortalidad.
- e)
Requiere la administración de una pauta antibiótica que cubra Gram positivos, Gram negativos y anaerobios.
Comentario: La fascitis necrotizante se presenta generalmente en pacientes con determinadas enfermedades basales predisponentes (DM, inmunodepresión, cirrosis, malnutrición, insuficiencia vascular periférica, insuficiencia renal). Aunque a veces no sea evidente, en muchas ocasiones es posible identificar una puerta de entrada (lesiones cutáneas, traumatismos, procedimientos quirúrgicos, etc.).
3. ¿Cuál de las siguientes afirmaciones le parece acertada?- a)
La mayor parte de casos de púrpura fulminans se producen en el contexto de enfermedades sistémicas.
- b)
La pustulosis exantemática aguda y el síndrome de hipersensibilidad inducido por fármacos pueden ocasionar una insuficiencia cutánea aguda.
- c)
Los linfomas cutáneos constituyen la causa más frecuente de eritrodermia.
- d)
Un exantema máculo papuloso que aparece 3 días después de haber iniciado tratamiento con carbamazepina sugiere un síndrome de hipersensibilidad inducido por fármacos.
- e)
El síndrome de la piel escaldada estafilocócica afecta mayoritariamente a pacientes mayores de 50 años.
Comentario: La insuficiencia cutánea aguda implica un grado severo de afectación cutánea que ha producido alteración de las funciones básicas de la piel. Dermatosis de diversa etiología pueden ocasionar esta situación. Entre las que con mayor frecuencia la producen se encuentran las toxicodermias, las enfermedades ampollosas y la psoriasis.
4. ¿Cuál de los siguientes hallazgos no forma parte del conjunto de criterios diagnósticos de un síndrome del shock tóxico estafilocócico?- a)
Trombopenia
- b)
Exantema maculoso difuso
- c)
Fiebre
- d)
Frecuencia respiratoria superior a 25 respiraciones por minuto.
- e)
Alteraciones del nivel de consciencia.
Comentario: El diagnóstico del SST estafilocócico incluye criterios generales (fiebre e hipotensión), cutáneos (exantema maculoso y descamación), sistémicos y de laboratorio. Se establece de forma definitiva cuando se cumplen los 6 criterios existentes y de forma probable cuando únicamente se cumplen 5.
5. ¿Cuál de las siguientes le parece la situación de menor gravedad?- a)
Presencia de lesiones ampollosas de contenido hemático asentadas en una zona de eritema e induración en la pierna derecha de una paciente diabética.
- b)
Lesiones maculosas eritematosas generalizadas con pequeñas zonas de despegamiento epidérmico en el tronco de un paciente a tratamiento con anticonvulsivantes.
- c)
Eritema difuso y numerosas pústulas diminutas, más acentuadas en los pliegues y acompañadas de prurito, edema facial y fiebre en un paciente a tratamiento previo con amoxicilina.
- d)
Edema, induración y lesiones purpúricas acompañadas de trombocitopenia, anemia e hipofibrinogenemia en un paciente con diagnóstico previo de hemangioendotelioma kaposiforme.
- e)
Exantema maculoso que se inicia en el tronco de una paciente 48h después de una laparotomía exploratoria y se acompaña de fiebre, vómitos e hipotensión.
Comentario: La fascitis necrotizante (a), la NET (b), el fenómeno de Kassabach-Merrit (d) y el síndrome del shock tóxico estafilocócico (e) son situaciones severas que presentan una mortalidad significativa. La pustulosis exantemática aguda (c) presenta una mortalidad muy inferior (en torno al 2%). Las lesiones suelen resolverse en unos días tras la suspensión del fármaco, dando paso a una descamación característica.