





Puntos clave
- •
La dermatomiositis (DM) es una entidad con lesiones cutáneas características y afectación muscular variable, existiendo incluso formas amiopáticas, que se puede relacionar con otras afectaciones sistémicas.
- •
Esta enfermedad constituye la miopatía inflamatoria más frecuente en la infancia.
- •
La patogenia resulta mal conocida. Si bien cada vez se conocen más factores predisponentes, los aspectos autoinmunitarios todavía permanecen en estudio, detectándose en los últimos tiempos varios anticuerpos específicos.
- •
La relación DM/malignidad se conoce desde que se describió la enfermedad, produciéndose en alrededor del 25% de los casos, siendo frecuentes las mismas neoplasias que en la población general.
- •
Respecto al tratamiento, los corticoides sistémicos son el de primera elección para la DM clásica y los corticoides tópicos de elevada potencia para las lesiones cutáneas.
- •
Se pueden usar otros inmunosupresores para ahorrar dosis de corticoides, además de nuevas terapias como tratamientos biológicos, con poca experiencia a día de hoy.
La dermatomiositis (DM) es una miopatía inflamatoria idiopática, que posee una sintomatología cutánea característica1,2. En 1975, Bohan y Peter1 establecieron 5 criterios que se siguen usando ampliamente con el objetivo de diagnosticar y clasificar la DM y la polimiositis (PM). De éstos, 4 se refieren a sintomatología muscular: debilidad simétrica, proximal y progresiva, aumento de las enzimas musculares, un electromiograma anormal y una biopsia muscular anormal. El quinto, que es el que diferencia la DM de la PM, se refiere a una sintomatología cutánea compatible.
Con estos criterios, se establecieron 5 tipos de miositis: la DM, la PM, la DM juvenil, la miositis con cáncer y la miositis superpuesta a otras enfermedades del tejido conjuntivo-vascular. Posteriormente se reconoció otro subtipo, la miositis con cuerpos de inclusión3 (MCI), simplificándose la miopatía inflamatoria idiopática en 3 grupos subtipos: la DM, la PM y la miositis con cuerpos de inclusión.
A su vez, de la DM se distinguen 3 subtipos (tabla 1). La DM clásica, cuando las lesiones cutáneas se manifiestan juntamente con debilidad muscular; la DM amiopática (DMA) o DM sine miositis, cuando la dermatitis no se asocia a sintomatología muscular durante al menos 6 meses después de la aparición de las lesiones cutáneas4,5, y la DM juvenil, cuando aparece en pacientes menores de 18 años. Ahora bien, la existencia de pacientes con lesiones cutáneas pero sin clínica muscular se conoce ya desde el inicio, pero cuando estos pacientes presentan alguna alteración en la biopsia muscular, en la resonancia magnética o en la ecografía muscular, a pesar de la normalidad en la sintomatología muscular y en las pruebas de laboratorio, se dice que presentan una DM hipomiopática (DMH)6. Esta forma puede progresar a un proceso generalizado, que requiera en un futuro un tratamiento sistémico. Por tanto, ante un paciente con clínica cutánea de DM y enzimas normales, debemos realizar otras pruebas para descartar una forma hipomiopática2.
Clasificación de la dermatomiositis
| Tipo | Características |
| DM clásica | Manifestaciones cutáneas compatibles con dermatomiositis con evidencia de debilidad muscular proximal durante los 6 primeros meses desde la afectación cutánea |
| DM clínicamente amiopática | Engloba las 2 formas siguientes |
| DM hipomiopática | Manifestaciones cutáneas compatibles con dermatomiositis durante 6 meses sin evidencia clínica o de laboratorio de miopatía |
| DM hipomiopática | Manifestaciones cutáneas compatibles con dermatomiositis sin debilidad muscular subjetiva durante más de 6 meses pero con evidencias de miositis subclínica en la biopsia muscular, elecromiograma o anormalidad en los enzimas musculares |
| DM juvenil | Dermatomiositis de cualquier tipo que aparece antes de los 18 años |
DM: dermatomiositis.
De Bendewald MJ et al7.
La DM clínicamente amiopática suele representar entre un 10 y un 30% del total de DM7,8 correspondiendo el resto a la DM clásica.
Las formas de DM juvenil son raras.
Lesiones cutáneasÚnicamente hay dos lesiones características, casi patognomónicas, de esta entidad: el eritema en heliotropo y las pápulas de Gottron2,9, siendo el resto de lesiones inespecíficas.
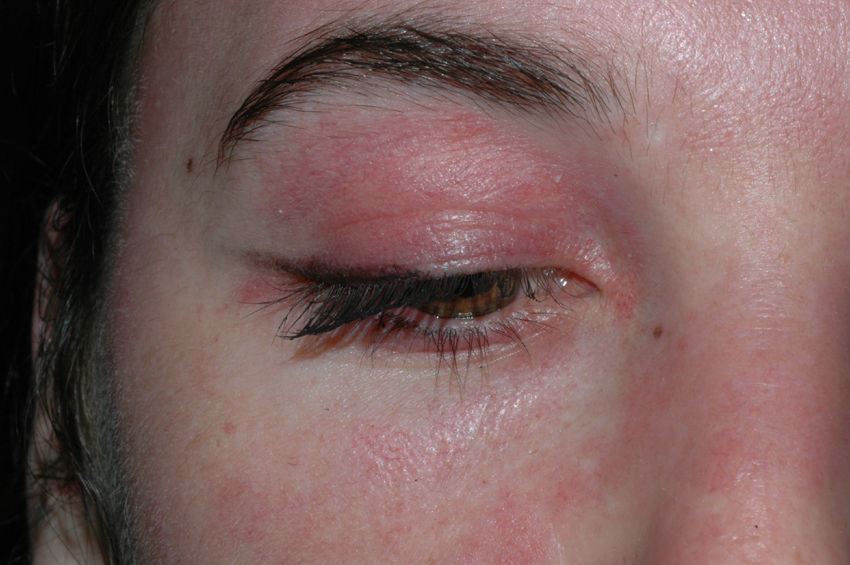
El eritema en heliotropo es un exantema eritematovioláceo que afecta a la piel periorbicular de forma simétrica y puede o no acompañarse de edema (fig. 1). Está causado, probablemente, por la dilatación venosa del músculo estriado inflamado, que se ve a través de la piel de los párpados9. En otras ocasiones, lo único que se aprecia es una coloración violácea leve del borde libre del párpado.

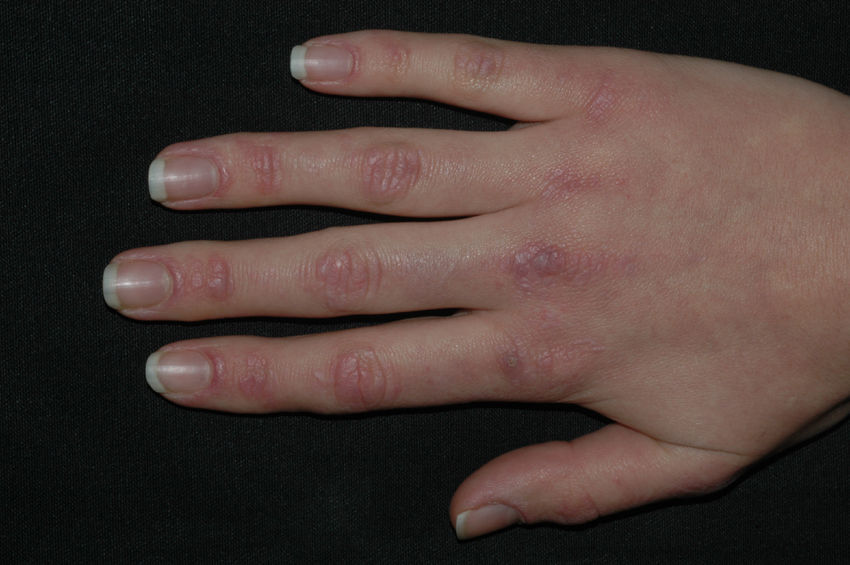
Las pápulas de Gottron (fig. 2) son pápulas o placas eritematovioláceas, ligeramente sobreelevadas y descamativas, que recubren las prominencias óseas. Las que se afectan más frecuentemente son las articulaciones metacarpofalángicas e interfalángicas proximales y distales, planteando el diagnóstico diferencial con lupus eritematoso o liquen plano. Es común la presencia de telangiectasias entre las lesiones. En otras ocasiones se afectan los codos, las rodillas o los pies. Se habla de signo de Gottron cuando únicamente se aprecian máculas violáceas en las mismas localizaciones.

En la biopsia cutánea de las lesiones patognomónicas de DM encontramos un infiltrado inflamatorio en la unión dermoepidérmica con una pobre vacuolización de la capa basal. También se observa un infiltrado dérmico perivascular formado por linfocitos T activados10, lo que hace que en ocasiones sea muy difícil de diferenciar del lupus eritematoso.
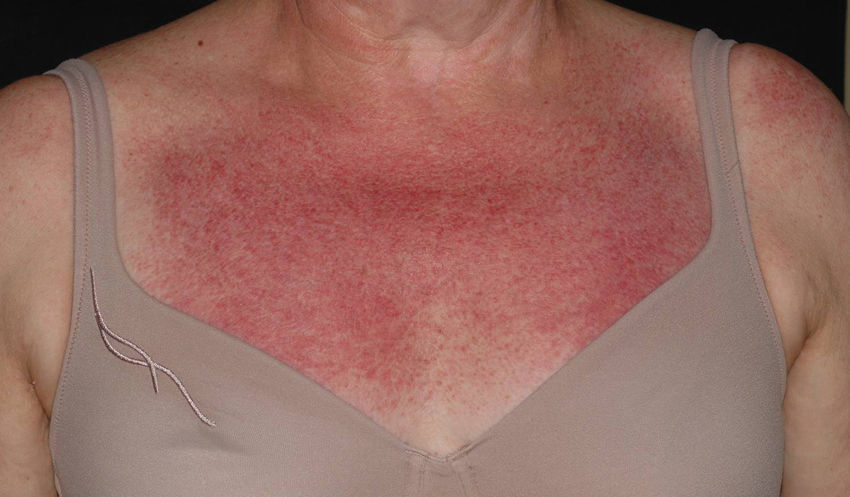
Entre las lesiones inespecíficas, cabe destacar un eritema malar fotosensible, muy parecido al del lupus eritematoso y poiquilodermia, consistente en atrofia, hiperpigmentación e hipopigmentación y telangiectasias, que se localiza, sobre todo, en la «V» torácica, recibiendo el nombre de «signo del chal» (fig. 3). También podemos encontrar un eritema violáceo en las superficies extensoras de las extremidades, hipertrofia de las cutículas, telangiectasias periungueales y alopecia no cicatrizal11. En la mucosa oral, en ocasiones puede apreciarse eritema, hemorragias, vesículas, aftas y leucoqueratosis12. Las telangiectasias gingivales13 y los angioqueratomas14 son manifestaciones muy raras de la forma juvenil de la enfermedad, pero igualmente descritas, lo mismo que las dermatosis Sweet-like en la edad adulta15.
Afectación muscular
Se presenta en forma de debilidad muscular proximal, normalmente simétrica, de instauración lenta, que puede ser de semanas a meses. Los pacientes describen una marcada incapacidad pera subir escaleras, levantarse de la silla, peinarse o afeitarse. Si realizamos biopsia muscular se observan, específicamente, miocitos atróficos y degenerados en una distribución claramente perifascicular, que se explican por la hipoxia producida por la destrucción de los capilares12. Otra característica es la inflamación perivascular, con infiltrado mixto de linfocitos B y linfocitos CD416. Entre un 15% y la mitad de los casos de DM se acompañan de disfagia, bien proximal por la afectación del músculo estriado, bien distal por afectación del músculo liso. En este último caso, de peor pronóstico, debe descartarse una superposición con esclerodermia2. También puede afectarse la musculatura estriada de la faringe, que da lugar a una disfagia alta17, y la de los músculos estriados intercostales, que puede conducir a disnea grave y puede precisar intubación. Entre un 15 y un 30% de los pacientes desarrollan enfermedad pulmonar, en forma de neumonitis intersticial. Puede deberse a la hipoventilación producida por la afectación muscular o por aspiración en pacientes con disfagia, ya que se ha visto mayor frecuencia de neumonitis en pacientes con disfunción esofágica9. Esta afectación pulmonar también es un marcador de mal pronóstico.
Dermatomiositis juvenilComo ya se ha comentado en la introducción, la DM juvenil es aquella que afecta a pacientes por debajo de los 18 años de edad. Aunque es una enfermedad rara, se estima que constituye un 85% de las miopatías inflamatorias idiopáticas de la infancia18. Las cifras de incidencia en Estados Unidos y el Reino Unido son de alrededor de 2-3 por millón al año19,20, con cierto predomino del sexo masculino 2,3:1 en todo el mundo19,21. La edad más frecuente de presentación se sitúa por debajo de 7 años22, con cifras incluso más elevadas por debajo de los 4 años23. La DM juvenil se considera una vasculopatía sistémica cuya sintomatología es idéntica a la forma del adulto. A veces se diagnostica tarde, ya que según la edad del paciente, todavía no realiza actividades que pongan de manifiesto su debilidad muscular ni puede comunicarlo en caso de presentarla. Su etiopatogenia es superponible a la del adulto, con los mismos factores predisponentes e iguales alteraciones autoinmunitarias (véase más adelante). A diferencia de la forma del adulto, no se asocia con aumento de riesgo de presentar cáncer, y las manifestaciones pulmonares son menos frecuentes24. En cambio, se produce la calcinosis hasta en un 40% de los casos, una tasa mucho mayor que en la forma del adulto.
Afectación sistémicaEs frecuente encontrar artralgias matutinas o incluso artritis en 25% de los pacientes, lo que hace evidente que la DM es un trastorno multisistémico25. Tanto la afectación pulmonar como la cardiaca son signos de mal pronóstico. La afectación cardiaca puede aparecer hasta en un 50% de los pacientes, aunque sólo se manifiesta en unos pocos, en forma de alteraciones en la conducción, insuficiencia cardiaca, pericarditis y valvulopatía9.
La calcinosis cutánea y muscular26 es frecuente en la forma juvenil de la DM (40%), pero inusual en el adulto, y se asocia a autoanticuerpos contra una proteína de 140kD27. Suele manifestarse en forma de nódulos blanco-amarillentos sobre las prominencias óseas2, que aunque son asintomáticos, pueden infectarse o producir deformidades en el hueso subyacente.
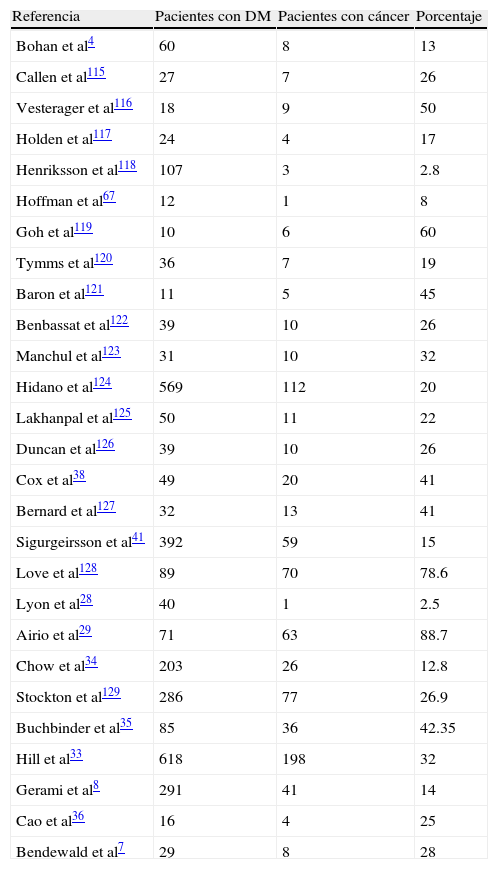
Cáncer asociado a dermatomiositisLa asociación de DM con la presencia de neoplasias internas se conoce desde la descripción de la enfermedad. El primer estudio de esta asociación, realizado por Bohan y Peter después de postular sus propios criterios, data de 1977. En él se comprobó una mayor incidencia de cáncer interno entre los pacientes con DM respecto a los que presentan PM4. Desde entonces se han publicado múltiples series en las que se analiza la incidencia de cáncer en estos pacientes (tabla 2). De estos estudios se deduce que la tasa de asociación entre DM y cáncer es muy variable, entre el 2,5% de la serie de Lyon et al28 y el 88,7% encontrado por Airio et al29, aunque por lo general se considera que esta asociación se produce entre el 20-25% de los pacientes con DM9,30, apareciendo pocos meses antes o después o coincidiendo con la sintomatología de DM. En cambio, si se asocia con PM, la neoplasia siempre se pone de manifiesto después de la aparición de los signos de inflamación muscular31.
Asociación entre dermatomiositis y cáncer
| Referencia | Pacientes con DM | Pacientes con cáncer | Porcentaje |
| Bohan et al4 | 60 | 8 | 13 |
| Callen et al115 | 27 | 7 | 26 |
| Vesterager et al116 | 18 | 9 | 50 |
| Holden et al117 | 24 | 4 | 17 |
| Henriksson et al118 | 107 | 3 | 2.8 |
| Hoffman et al67 | 12 | 1 | 8 |
| Goh et al119 | 10 | 6 | 60 |
| Tymms et al120 | 36 | 7 | 19 |
| Baron et al121 | 11 | 5 | 45 |
| Benbassat et al122 | 39 | 10 | 26 |
| Manchul et al123 | 31 | 10 | 32 |
| Hidano et al124 | 569 | 112 | 20 |
| Lakhanpal et al125 | 50 | 11 | 22 |
| Duncan et al126 | 39 | 10 | 26 |
| Cox et al38 | 49 | 20 | 41 |
| Bernard et al127 | 32 | 13 | 41 |
| Sigurgeirsson et al41 | 392 | 59 | 15 |
| Love et al128 | 89 | 70 | 78.6 |
| Lyon et al28 | 40 | 1 | 2.5 |
| Airio et al29 | 71 | 63 | 88.7 |
| Chow et al34 | 203 | 26 | 12.8 |
| Stockton et al129 | 286 | 77 | 26.9 |
| Buchbinder et al35 | 85 | 36 | 42.35 |
| Hill et al33 | 618 | 198 | 32 |
| Gerami et al8 | 291 | 41 | 14 |
| Cao et al36 | 16 | 4 | 25 |
| Bendewald et al7 | 29 | 8 | 28 |
Todos los estudios realizados demuestran que los pacientes con DM tienen mayor riesgo de presentar cáncer que los pacientes con PM y que ambas enfermedades tienen mayor incidencia de neoplasias que la población general29,32–34. Este riesgo sigue siendo elevado respecto a la población general durante varios años después del diagnóstico de la DM. Buchbinder et al35 establecen este periodo de tiempo de hasta 5 años, pero en 2 estudios se encuentran neoplasias incluso después de pasar este intervalo33–35. En un estudio se propone una definición de temporalidad de 3 años para que se pueda asociar malignidad y DM4. Viendo las observaciones epidemiológicas descritas en los diferentes estudios, esta cifra es la que debería ser razonablemente definitoria31, descendiendo el riesgo pasados estos años. Aunque la forma clásica de DM sea la que más se asocia a malignidad, los pacientes con subtipos clínicamente amiopáticos también tienen incrementado el riesgo de sufrir cáncer8, así, recientemente Gerami et al8 encuentran un 14% de asociación y Cao et al36 un 25%.
Se ha visto que la probabilidad de neoplasia aumenta con la edad; cuando ésta aparece es común que sea en pacientes mayores de 50 años (siendo entre 50 y 60 años el periodo con más incidencia31), pero no se han realizado estudios que demuestren la independencia de estas variables. Raramente se producen neoplasias en pacientes con DM juvenil. Se considera que no hay relación entre esta forma clínica de DM y el cáncer.
Clásicamente, el tumor que más representado está en estos pacientes es el cáncer de colon31. En mujeres, las neoplasias de ovario ocupan la primera posición, siendo Barnes et al37 los que antes apreciaron esta asociación. Esta hipótesis se vio reforzada por los estudios de Cox et al38, Scaling et al39, Verducci et al40 y Sigurgeirsson et al41, en los que se concluyó que este tipo de cáncer era mayoritario respecto al resto de tumores, aunque estudios escandinavos más recientes encuentran un aumento en la incidencia de cáncer de pulmón, páncreas, estómago, colorrectal y linfoma no hodgkiniano33 tanto en hombres como en mujeres. En cambio, en países asiáticos, el carcinoma que predomina es el nasofaríngeo32,42 lo que indica que las neoplasias asociadas a DM son las mismas que las que aparecen con mayor frecuencia en la población general por grupo de edad, sexo y raza.
Por tanto, las exploraciones que se deberían realizar a todo paciente con DM para la detección de neoplasias deberían ser: hemograma completo, velocidad de sedimentación globular (VSG), bioquímica general, marcadores tumorales (sobre todo CA125 y CA19.943, radiografía de tórax, citología urinaria, prueba de sangre oculta en heces o colonoscopia en caso de sospecha de cáncer de colon, tomografía computarizada [TC] torácica y ecografía abdominal [o TC en su defecto]). Además, en mujeres, se deberían añadir una mamografía, TC o ecografía pélvica y exploración ginecológica.
Además, por su valor predictivo respecto a la aparición de cáncer (sensibilidad del 50% y especificidad del 96%, véase más adelante), sería importante determinar los niveles de autoanticuerpo anti-155/140 en todos los pacientes con DM, realizando exámenes de detección más exhaustivos en caso de positividad. Por el contrario, se ha confirmado el factor protector del autoanticuerpo anti-Jo-1, cuando este se presenta de forma aislada (sin asociarse a anti-155/140)31 en pacientes con CADM.
PatogeniaEl mecanismo autoinmunitario que conduce a la etiología de la DM no se conoce todavía de forma completa debido a su complejidad. Se sabe que hay individuos con más predisposición a esta enfermedad, como los que poseen ciertos alelos de HLA (en especial los asociados con el haplotipo 8.1 AH12, que se relaciona con la aparición de anticuerpos tanto asociados como específicos de miositis), con el fenotipo Gm 3 25 3,13 de la cadena pesada de las gammaglobulinas44 y el polimorfismo -308A del factor de necrosis tumoral (FTN)12,45,46.
Además, ciertos medicamentos inducen la aparición de la enfermedad o exacerban la sintomatología de ésta. Entre los que más frecuentemente la desencadenan se hallan la hidroxiurea47,48 y los antagonistas del FNT49–51. Otros medicamentos que también se han relacionado con la aparición de DM son la quinidina, los antiinflamatorios no esteroideos y la penicilamida2.
Parece ser que la importancia del FNT-¿ en la DM es crucial. Se cree que la luz ultravioleta, que es un potencial desencadenante en las lesiones cutáneas de DM, libera FNT-¿ de los queratinocitos y los fibroblastos, además de producir apoptosis de éstos, lo cual estimularía más aún la producción de FNT-¿52. Una vez liberada, esta citocina provocaría la maduración de las células presentadoras de antígenos (CPA), aumentando la expresión del complejo mayor de histocompatibilidad (CMH) tipo I, lo que activaría la citotoxicidad de las células natural killer (NK) y promovería la activación y supervivencia de los linfocitos T45. Parece ser que los linfocitos B tienen también una importante función. Los linfocitos B CD4+, que mediante inmunohistoquímica se ha podido identificar que son CD19+ y CD20+53, se hallan presentes en las regiones perivasculares y perifasciculares, envolviendo al linfocito T y produciendo inmunoglobulinas que junto con el complemento forman complejos que se depositan en estas zonas54. De los anticuerpos producidos, ya hablaremos más en detalle más adelante.
Una nueva técnica para distinguir la DM del resto de miopatías inflamatorias idiopáticas es la descrita por Jain et al55, que consiste en la detección, mediante inmunohistoquímica, de un complejo de ataque a la membrana (CAM) en la biopsia muscular. La sensibilidad de esta prueba es del 80,9% y la especificidad es del 85% a la hora de discernir entre DM y las demás MII, pero todavía se tiene que estudiar con más detenimiento el rol de este complejo en la patogenia de la DM.
Dermatomiositis y autoanticuerposAnteriormente hemos descrito la fuerte asociación entre DM y autoinmunidad. Este hecho se ve reforzado porque entre el 50 y el 80% de los pacientes presentan altos valores de autoanticuerpos circulantes56,57.
Los anticuerpos presentes en la miositis se dividen en 2 subtipos: los autoanticuerpos asociados a miositis (MAA, del inglés myositis-associated autoantibodies) y los autoanticuerpos específicos de miositis (MSA, del inglés myositis-specific autoantibodies).
Autoanticuerpos específicos de miositis (tabla 3)Todavía no hay consenso acerca de si la aparición de MSA en la DM se debe a un epifenómeno o si se asocia directamente con la patogenia de ésta. El hecho de que estos autoanticuerpos se dirijan contra proteínas intracelulares puede orientar a que ellos mismos tengan un papel importante en el mecanismo de la enfermedad, pero las hipótesis formuladas hasta hoy son dispares y ninguna está aceptada de manera definitiva.
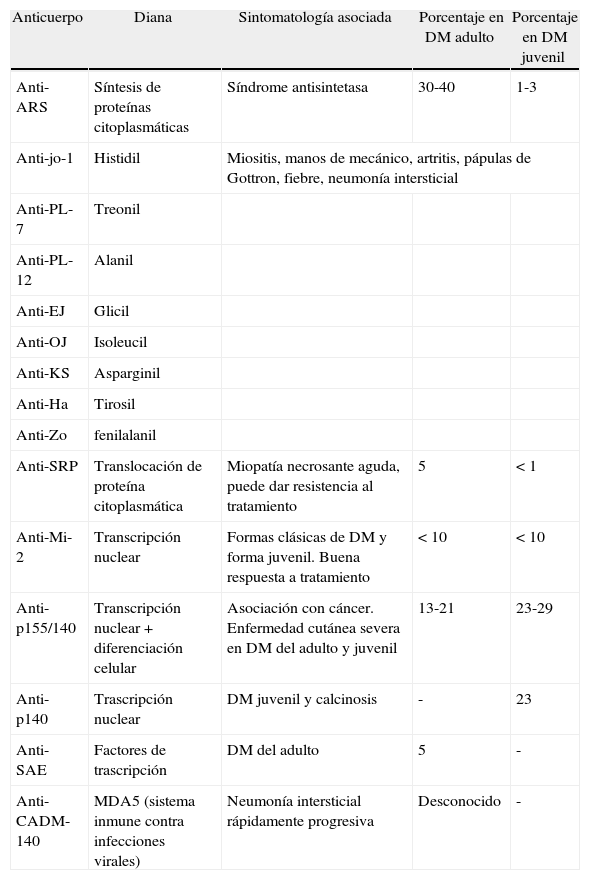
Clasificación de los anticuerpos específicos de miositis
| Anticuerpo | Diana | Sintomatología asociada | Porcentaje en DM adulto | Porcentaje en DM juvenil |
| Anti-ARS | Síntesis de proteínas citoplasmáticas | Síndrome antisintetasa | 30-40 | 1-3 |
| Anti-jo-1 | Histidil | Miositis, manos de mecánico, artritis, pápulas de Gottron, fiebre, neumonía intersticial | ||
| Anti-PL-7 | Treonil | |||
| Anti-PL-12 | Alanil | |||
| Anti-EJ | Glicil | |||
| Anti-OJ | Isoleucil | |||
| Anti-KS | Asparginil | |||
| Anti-Ha | Tirosil | |||
| Anti-Zo | fenilalanil | |||
| Anti-SRP | Translocación de proteína citoplasmática | Miopatía necrosante aguda, puede dar resistencia al tratamiento | 5 | < 1 |
| Anti-Mi-2 | Transcripción nuclear | Formas clásicas de DM y forma juvenil. Buena respuesta a tratamiento | < 10 | < 10 |
| Anti-p155/140 | Transcripción nuclear+diferenciación celular | Asociación con cáncer. Enfermedad cutánea severa en DM del adulto y juvenil | 13-21 | 23-29 |
| Anti-p140 | Trascripción nuclear | DM juvenil y calcinosis | - | 23 |
| Anti-SAE | Factores de trascripción | DM del adulto | 5 | - |
| Anti-CADM-140 | MDA5 (sistema inmune contra infecciones virales) | Neumonía intersticial rápidamente progresiva | Desconocido | - |
-: no datos aplicables.
De Gunawardena H et al57.
Los anticuerpos antiaminocil-tRNA sintetasa citoplasmática (ARS, del inglés aminoacyl-tRNA synthetase) son los más frecuentes en los pacientes adultos con miositis57. De ellos, el que predomina es el anti-jo-1 (antihistidil-tRNA sintetasa)56, que se encuentra aproximadamente en un 20% de los pacientes, observándose una cierta asociación entre sus niveles y las cifras de CK, intensidad de la miositis y enfermedad articular58. Los otros autoanticuerpos de este grupo son el PL-7 (antitreonil), PL-12 (antialanil), EJ (antiglicil), OJ (antiisoleucil), KS (antiasparginil), Ha (antitirosil) y Zo (antifenilalanil). Todos ellos pueden ser hallados en el síndrome antisintetasa, que abarca un amplio espectro de manifestaciones clínicas entre las que se incluyen la miositis, neumonitis intersticial, artritis no erosiva, fiebre y manos de mecánico.
Otro tipo de MSA relacionado con la DM es el SRP, dirigido contra un complejo ribonucleoproteico citoplasmático que regula la translocación de proteínas a través del retículo endoplasmático59.
Los anticuerpos anti-Mi-2 (proteína de una helicasa nuclear, que tiene un papel importante en la transcripción génica60) se detectan en pacientes con lesiones características de DM. El anticuerpo anti-p155/140 actúa contra esta proteína, que es una intermediaria en la trascripción de factor-1 gamma, implicada en la diferenciación celular. La importancia de este autoanticuerpo en la DM del adulto radica en su relación con la aparición de neoplasias asociadas. En cambio, en la forma juvenil de la DM, tal asociación no se ha evidenciado61. Aunque la presencia de anti-Jo-1 y anti-Mi-2 se ha relacionado con la aparición de neoplasias en pacientes con DM, en especial con adenocarcinomas de mama y pulmón62, se ha visto que únicamente el anticuerpo anti-p155/40 se asocia de manera significativa con la aparición de cáncer56, por lo que debería ser monitorizado para el diagnóstico precoz de neoplasias.
En la DM juvenil se han detectado autoanticuerpos anti-p140kDa27 que se asocian con la presencia de calcinosis57. Estos actúan contra la proteína NXP-2, un factor de trascripción situado en el núcleo. En el 2005, Sato et al63 describieron otro anticuerpo de la familia de los MSA descubierto en un grupo de pacientes japoneses con DM clínicamente amiopática, el anti-CADM140 (anti-clinically amyopathic dermatomyositis 140) dirigido contra el MDA5 (melanoma-differentiation-associated gene 5) que se asocia a enfermedad pulmonar rápidamente progresiva, pero nunca a neoplasias. El último MSA descrito es el anti-SAE, contra una enzima activadora/modificadora de ubiquitina, localizada tanto en el núcleo como en el citoplasma64.
Anticuerpos asociados a miositisEn este grupo están representados aquellos anticuerpos que se han encontrado en pacientes con DM, pero sin asociación específica con ésta. Inicialmente se describieron en pacientes con superposición entre DM y esclerodermia65. Aquí se incluyen los anticuerpos contra los antígenos PM-Scl, Ku, snRNP (small nuclear RNP), y los Ro citoplasmáticos, como el Ro60/SSA, La/SSB y Ro5231. No se han dedicado muchos estudios por su escasa importancia en relación con la DM y sus complicaciones.
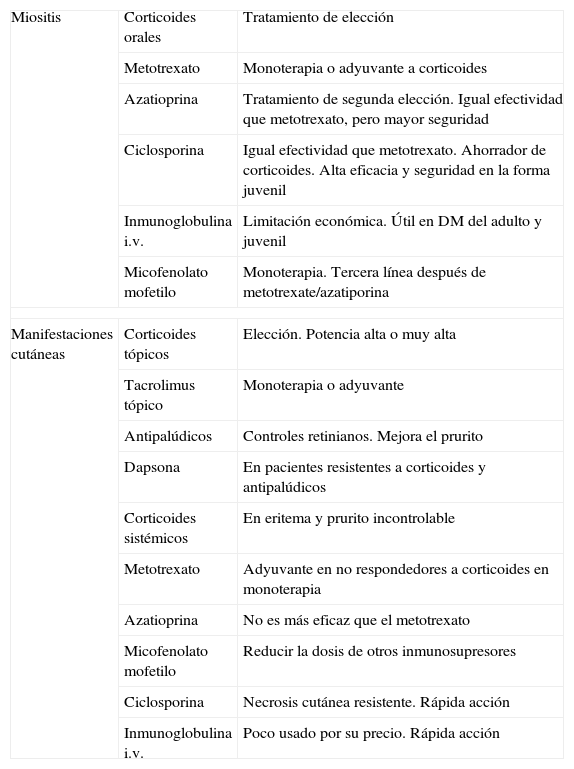
Tratamiento de la dermatomiositisEn este apartado valoraremos los diferentes tratamientos tanto para las manifestaciones musculares como para las lesiones cutáneas (tabla 4).
Tratamiento de la dermatomiositis
| Miositis | Corticoides orales | Tratamiento de elección |
| Metotrexato | Monoterapia o adyuvante a corticoides | |
| Azatioprina | Tratamiento de segunda elección. Igual efectividad que metotrexato, pero mayor seguridad | |
| Ciclosporina | Igual efectividad que metotrexato. Ahorrador de corticoides. Alta eficacia y seguridad en la forma juvenil | |
| Inmunoglobulina i.v. | Limitación económica. Útil en DM del adulto y juvenil | |
| Micofenolato mofetilo | Monoterapia. Tercera línea después de metotrexate/azatiporina | |
| Manifestaciones cutáneas | Corticoides tópicos | Elección. Potencia alta o muy alta |
| Tacrolimus tópico | Monoterapia o adyuvante | |
| Antipalúdicos | Controles retinianos. Mejora el prurito | |
| Dapsona | En pacientes resistentes a corticoides y antipalúdicos | |
| Corticoides sistémicos | En eritema y prurito incontrolable | |
| Metotrexato | Adyuvante en no respondedores a corticoides en monoterapia | |
| Azatioprina | No es más eficaz que el metotrexato | |
| Micofenolato mofetilo | Reducir la dosis de otros inmunosupresores | |
| Ciclosporina | Necrosis cutánea resistente. Rápida acción | |
| Inmunoglobulina i.v. | Poco usado por su precio. Rápida acción | |
Es el tratamiento de elección para esta manifestación clínica. La Academia Americana de Dermatología recomienda dosis iniciales de 0,5 a 1,5mg/kg/día hasta controlar los síntomas, y una vez conseguida la normalización de las enzimas musculares, reducirlos paulatinamente hasta la suspensión66. La respuesta a este tratamiento es muy variable, pero alrededor de un 90% de los pacientes mejoran y muchos pueden finalizarlo de forma completa en aproximadamente 2 o 3 años67,68. En la forma juvenil, aunque se había postulado dosis más elevadas (> 2mg/kg/día)69, no existe ningún estudio aleatorizado que lo avale, por lo que mayormente se administran dosis de 1mg/kg/día, seguido de un descenso durante 6 meses, con el objetivo de finalizar el tratamiento en 2 años66.
MetotrexatoPuede administrarse de forma oral, subcutánea o intramuscular70,71 a una dosis de entre 15 y 25mg a la semana72. Se considera el tratamiento de elección para las formas resistentes a los corticoides, y combinado con corticoides en los pacientes con miositis grave73,74. Con estas pautas mejoran hasta el 75% de los pacientes75,76. El metotrexato también es útil para aquellos pacientes con lesiones cutáneas resistentes a otros tratamientos66 a pesar de que la miositis esté bien controlada.
AzatioprinaSe utiliza en pacientes que no responden a los corticoides, con una mejora clínica del 57% al 75%75. Ya que se ha comprobado que la eficacia de la azatioprina es la misma que la del metotrexate, pero con un margen de seguridad mayor, debería de considerarse éste como tratamiento de segunda elección.
En la DM juvenil, la azatioprina se ha utilizado con éxito como ahorrador de corticoides77.
CiclosporinaNo se ha demostrado que la ciclosporina sea más efectiva que el metotrexato en el tratamiento de la miositis78, pero se ha visto que es útil en el tratamiento de la DM juvenil resistente79,80 y en la neumonía intersticial resistente a corticoides81. La ciclosporina debería de administrarse como ahorrador de prednisona en pacientes en los que esté contraindicado el uso de metotrexato o azatioprina.
Inmunoglobulina intravenosaSe han utilizado con éxito hasta en un 92% de pacientes con enfermedad resistente82,83 con efectos adversos muy bien tolerados. Con iguales resultados se ha administrado a pacientes con DM juvenil84,85. Su coste impide que este tratamiento se generaliza como alternativa en pacientes resistentes.
Micofenolato mofetilAdministrado en forma de monoterapia, sin necesidad de asociarse a corticoides, se ha visto que es uno de los tratamientos más efectivos, tanto para las lesiones cutáneas como para la miositis86–89. Podría considerarse como fármaco de segunda elección después del metotrexato/azatiporina, aunque podría favorecer la aparición de infecciones oportunistas90.
Otras terapiasEn los 2 estudios realizados hasta la fecha con rituximab se ha conseguido una mejora de la afectación muscular91,92, pero sin que se hayan observado cambios en las lesiones cutáneas. Es posible que este fármaco sea considerado en el futuro uno de los tratamientos de elección, sobre todo si se superan las limitaciones de coste y se conozca mejor su toxicidad.
En pacientes con DM resistente se han utilizado, en forma de monoterapia, fármacos anti-TNF-¿ como el etanercept o el infliximab con buenos resultados93–96. En cambio, con la plasmaféresis no ha obtenido beneficio alguno en el tratamiento de estos pacientes97.
En el tratamiento de la DM juvenil, Riley et al98 proponen la ciclofosfamida i.v. como alternativa exitosa y segura en pacientes que no responden a otras terapias, alcanzando sus pacientes una dosis acumulada de 4,6g/m2 de media en 6 meses.
En los pacientes afectados de miositis, además de estas consideraciones farmacológicas, cabe destacar la importancia de la rehabilitación muscular y el ejercicio físico, con la finalidad de evitar la atrofia muscular y las posteriores complicaciones y el soporte psicológico, en especial en los pacientes con DM juvenil20.
Manifestaciones cutáneasLa base para un buen control de las lesiones cutáneas de la DM es, fundamentalmente, la fotoprotección, ya que la fotosensibilidad ocurre aproximadamente en un 50% de los pacientes. Además, los tratamientos utilizados para estas manifestaciones son:
Corticoides tópicosPara controlar el picor y el eritema se deben utilizar corticoides de potencia alta o muy alta en las zonas donde no estén contraindicados99. En el cuero cabelludo deben usarse lociones, geles, emulsiones o espumas. En las lesiones hiperqueratósicas es preferible utilizarlos en oclusión100.
AntihistamínicosCon el fin de reducir el disconfort y el prurito, podemos recomendar el uso de antihistamínicos no sedantes durante el día, combinado con antihistamínicos sedantes antes de ir a dormir, para reducir las molestias nocturnas causadas por la inflamación72,100.
Tacrolimus tópicoEl tacrolimus tópico al 0,1% es una buena alternativa en el tratamiento de lesiones cutáneas resistentes. Se puede utilizar tanto en adultos como en niños, ya sea en monoterapia o como coadyuvante, porque se ha visto que mejora la clínica, aunque faltan estudios randomizados potentes que lo demuestren24.
AntipalúdicosSu efecto antiinflamatorio reside en la inhibición de la quimiotaxis de los neutrófilos, macrófagos y eosinófilos24. La hidroxicloroquina mejora las lesiones cutáneas y el prurito localizado en el cuero cabelludo101,102, aunque algunos pacientes ocasionalmente desarrollan una reacción cutánea pruriginosa dentro de las 3 primeras semanas de tratamiento103. Debe iniciarse a una dosis de 6mg/kg/día hasta alcanzar la respuesta clínica y reducirla posteriormente a dosis de mantenimiento de 3mg/kg/día durante un año, con controles retinianos periódicos, para reducir el riesgo de recurrencias100. El hecho de que la hidroxicloroquina no tenga efectos sobre la enfermedad muscular, sugiere que el mecanismo de afectación de la piel es diferente al del músculo.
La cloroquina es menos utilizada por su mayor riesgo de retinopatía. Debe administrarse una dosis menor de 3mg/kg/día. Además, se han descrito algunos casos de miositis inducida por cloroquina, que debe sospecharse en pacientes en los que empeora su debilidad muscular en ausencia de elevación enzimática o alteraciones en el electromiograma24.
DapsonaActúa inhibiendo los linfocitos y el bloqueando la vía alternativa del complemento. Se debe iniciar a una dosis de 50mg/día hasta alcanzar la mejora clínica o una dosis máxima de hasta 300mg/día. Se utiliza como tratamiento en pacientes con lesiones cutáneas resistentes a corticoides y antipalúdicos.
Corticoides sistémicosPueden utilizarse en casos de eritema y prurito intratables, a dosis de 1mg/kg/día por vía oral, o bien en pulsos intravenosos de metilprednisolona (30mg/kg/día durante 3 días consecutivos104).
MetotrexatoSe considera como primera línea de tratamiento adyuvante en pacientes que no responden a corticoides en monoterapia. La dosis recomendada es de 7,5 a 14,5mg/semana105.
AzatioprinaSe utiliza como fármaco ahorrador de corticoides sistémicos en la terapia de las lesiones cutáneas, pero su eficacia es menor que la del metotrexato106.
Micofenolato mofetiloAdministrado en dosis de 500mg/día durante 4-8 semanas mejora las lesiones cutáneas89. Los efectos adversos más comunes son las molestias gastrointestinales y las alteraciones hematológicas. Se considera útil en pacientes en tratamiento con inmunosupresores para reducir dosis de éstos.
CiclofosfamidaNo se ha evidenciado mejora de las lesiones cutáneas con este tratamiento72, pero es útil en el tratamiento de la enfermedad pulmonar intersticial asociada a DM107.
CiclosporinaLa ventaja de la ciclosporina es su rapidez de acción en la mejora de la sintomatología cutánea, que se observa a partir de la segunda semana de tratamiento. Se recomienda una duración de uno a dos años, utilizando la mínima dosis de mantenimiento posible. Es muy útil en pacientes con necrosis cutánea resistente108,109.
Inmunoglobulina intravenosaPor su elevado coste, esta alternativa debe reservarse para pacientes que no respondan a ninguno de los tratamientos propuestos anteriormente24. La respuesta terapéutica es rápida, observándose a las 1-2 semanas del inicio del tratamiento110.
Otros tratamientosOtras opciones terapéuticas son el rituximab y los inhibidores del TNF-¿, con buenas perspectivas de futuro. Levine91 y Silverman et al111 han demostrado resultados prometedores con rituximab (anticuerpo monoclonal anti-CD20) en pacientes con afectación cutánea, pulmonar y muscular resistentes al tratamiento habitual.
Se ha comprobado que el infliximab es eficaz en pacientes con DM amiopática y lesiones cutáneas, administrándose 3 infusiones de 10mg/kg separadas por 2 semanas112. El etanercept en dosis estándar también ha sido estudiado con buenos resultados para el tratamiento de estas lesiones93.
La plasmaféresis no ha demostrado efectividad en el tratamiento de las lesiones cutáneas.
Tratamiento de la calcinosisLa warfarina administrada a 1mg/día puede ser útil en la calcinosis incipiente. El diltiazem en altas dosis (360mg/día)113, los bifosfonatos y el probenecid114 también se utilizan para tratar esta complicación. La cirugía se reserva para cuando el tratamiento es inefectivo o las lesiones son muy dolorosas24.
ConclusionesLa dermatomiositis es una entidad con lesiones cutáneas características y afectación muscular variable, existiendo incluso formas amiopáticas, que se puede relacionar con otras afectaciones sistémicas. Esta enfermedad constituye la miopatía inflamatoria más frecuente en la infancia. La patogenia resulta mal conocida. Si bien cada vez se conocen más factores predisponentes, los aspectos autoinmunes todavía permanecen en estudio, detectándose en los últimos tiempos varios anticuerpos específicos.
La relación DM/malignidad se conoce desde que se describió la enfermedad. A pesar de la variabilidad entre los diferentes estudios, se considera esta asociación se produce en alrededor del 25% de los casos, siendo frecuentes las mismas neoplasias que en la población general. Respecto al tratamiento, los corticoides sistémicos son de primera elección para la DM clásica y los corticoides tópicos de elevada potencia para las lesiones cutáneas. Se pueden usar otros inmunosupresores para ahorrar dosis de corticoides, además de nuevas terapias como tratamientos biológicos, con poca experiencia a día de hoy.