




La Enfermedad Granulomatosa Crónica, es una Inmunodeficiencia Primaria (IDP), clasificada dentro de los defectos funcionales de fagocitos, asociada a mutaciones en el complejo multiprotéico NADPH oxidasa, las cuales se asocian con el patrón de herencia de la enfermedad, siendo más frecuente los casos ligados al cromosoma X con una frecuencia del 60%, en donde es importante encontrar a las madres portadoras de la enfermedad. Las manifestaciones clínicas de esta enfermedad son infecciones recurrentes y severas generadas por bacterias catalasa positivas, además de levaduras. Los hallazgos clínicos más frecuentes son infecciones severas en piel, adenitis, hepatoesplenomegalia, neumonía recurrente, formación de granulomas y en aproximadamente un 40% de los pacientes BCGitis.
El diagnóstico de la enfermedad granulomatosa crónica se ha realizado tradicionalmente con la técnica colorimétrica de nitro azul tetrazolium (NBT). Sin embargo, actualmente se dispone del ensayo de Dihidrorodamina 1,2,3 el cual se caracteriza por ser más sensible y específico además de permitir la detección del fenómeno de Lyonización en madres portadoras de la enfermedad como la actividad residual de la enzima.
El objetivo de este estudio es describir el reporte de un caso clínico de EGC y la importancia del ensayo de Dihidrorodamina 1,2,3 por citometrìa de flujo, como apoyo en el diagnóstico de la EGC. Se trata de un estudio de reporte de caso, descriptivo, en el cual se realizó una búsqueda en la literatura científica, encontrando una correlación con los hallazgos clínicos y de laboratorio del paciente y su progenitora. Como resultado final del estudio, se observó una disminución severa en la producción de especies reactivas del oxígeno (EROs) en el paciente, además del fenómeno de Lyonización en su progenitora.
Frente a la sospecha clínica de alteración funcional de fagocitos, la prueba de oxidación de Dihidrorodamina permite detectar la alteración en la producción especies reactivas del oxígeno (EROs) como el fenómeno de Lyonización.
Chronic granulomatous disease is a primary immunodeficiency (PID) classified within the phagocytes functional defects associated with mutations in the multi-protein enzyme complex, NADPH oxidase, which are associated with the pattern of inheritance of the disease. X-linked cases have a frequency of 60%, which makes it important to find surrogates of the disease. The clinical manifestations of this disease are recurrent, and with severe infections generated by catalase positive bacteria, as well as yeasts. The most common clinical findings are severe skin infections, lymphadenitis, hepatosplenomegaly, recurrent pneumonia, and granuloma formation in approximately 40% of patients with BCGitis.
The diagnosis of chronic granulomatous disease has traditionally been made with the colorimetric nitro blue tetrazolium (NBT) technique. However, the currently available 1,2,3 dihydrorhodamine assay, which has shown to be more sensitive and specific, in addition to enable detection of the lyonisation phenomenon in mothers carrying the disease, as well as the residual activity of the enzyme.
The aim of this study was to report a case of CGD, and the importance of 1,2,3 dihydrorhodamine test by flow cytometry, as support in its diagnosis. It is a descriptive study case report, and includes a search in the scientific literature, and finding a correlation with clinical and laboratory findings of the patient and the mother.
The end result of the study, a severe decrease was observed in the production of reactive oxygen species (ROS) in the patient, in addition to the lyonisation phenomenon in the mother.
Faced with clinical suspicion of functional impairment of phagocytes, the dihydrorhodamine oxidation test can detect the alteration in the production of reactive oxygen species (ROS) as well as the lyonisation phenomenon.
La Enfermedad Granulomatosa Crónica (EGC), es una Inmunodeficiencia Primaria (IDP), clasificada dentro de los defectos funcionales de fagocitos, asociada a mutaciones que afectan el complejo multiprotéico NADPH oxidasa, con la consecuente disminución en la producción de las Especies Reactivas del metabolismo del Oxígeno (EROs), incrementando la susceptibilidad a infecciones recurrentes. Esta enfermedad se manifiesta con mayor predominio en la infancia con una prevalencia de 1 caso por cada 200.000 mil nacidos vivos. Las manifestaciones clínicas son infecciones recurrentes y severas por bacterias catalasa positivas, además de levaduras1–3. Los hallazgos clínicos más frecuentes son, linfadenopatía marcada, hepatoesplenomegalia, neumonía recurrente, además de otras manifestaciones como conjuntivitis, sinusitis, dermatitis, estomatitis ulcerativas, diarrea crónica, obstrucción intestinal y osteomielitis supurativa4–6.
El primer hallazgo clínico suele estar constituido por una adenitis localizada en el área de aplicación de la vacuna BCG, también llamada BCGitis, o BCGosis en su forma diseminada, o la presencia de adenopatías supurativas que se localizan con mayor frecuencia en el cuello6–9. Generalmente las infecciones no son controladas por la actividad de células fagocíticas, motivo por el cual se pueden formar granulomas, en los cuales se encuentran macrófagos activados dando lugar a obstrucciones uretrales, pilóricas y esofágicas. Las anormalidades en el tracto urinario son relativamente frecuentes, por lo cual es importante sospechar EGC en niños que cursen con síntomas o signos de obstrucción de la vía urinaria7–10.
La NADPH oxidasa es un complejo multiprotéico que se encuentra presente en la membrana celular de células fagociticas como el neutrófilo, monocito y eosinófilo. Esta enzima en su estado basal se encuentra conformada por 2 proteínas expresadas constitutivamente en la membrana (gp91phox y p22phox), las cuales conforman el centro activo enzimático o flavocitocromo b558, además de 4 proteínas citosólicas (p40phox, p47phox, p67phox y Rac)11–14. Durante el proceso de activación de este complejo, las proteínas citosólicas fosforiladas por acción de la proteína cinasa C (PKC), se traslocan a la membrana e interactúan con el flavocitocromo, conformando la forma activa de la enzima, la cual transforma el oxígeno molecular en EROs, tales como el anión superóxido, peróxido de hidrógeno y ácido hipocloroso, los cuales son responsables de la oxidación de proteínas, lípidos, carbohidratos y ácidos nucleicos presentes en agentes infecciosos, dentro de un proceso llamado estallido respiratorio7–11.
Para el diagnóstico microbiológico es importante el aislamiento de agentes infecciosos extracelulares, bacterias catalasa positiva como Staphylococcus aureus, Pseudomona aeruginosa, Pantoea aglomerans, Serratia marcenses, bacilos gram negativos como E. coli, Salmonella sp y otros como Nocardia sp., Además de hongos como Candida sp y Aspergillus sp9–12.
El estudio inicial de los pacientes con sospecha clínica de EGC se puede realizar con un método sencillo y económico como la reducción del NBT (nitro azul tetrazolium), el cual se fundamenta en la reducción del NBT soluble de color amarillo, en azul de formazán insoluble, por acción del anión superóxido, bajo el estímulo con Lipopolisacarido (LPS) bacteriano o con PMA.
Dentro de las limitaciones de esta prueba se puede citar su baja sensibilidad, además de su interpretación subjetiva y, de no medir defectos parciales de la NADPH oxidasa, en los que se observa una actividad residual de la enzima en las muestras estimuladas con PMA, como la detección del fenómeno de Lyonización15–18.
El ensayo con Dihidrorodamina se fundamenta en la oxidación de Dihidrorhodamina 1,2,3 a Rodamina1, 2, 3; una molécula fluorescente, por acción del peróxido de hidrogeno (H2O2) y otros EROs presentes en polimorfonucleares neutrófilos y monocitos, al realizar el estímulo con PMA. Esta prueba diagnóstica tiene ventajas en comparación con la reducción del NBT, permite la medición del estallido respiratorio en neutrófilos y monocitos por separado, además de detectar la presencia de actividad residual de la enzima, y detectar el fenómeno de Lyonización en madres portadoras de la enfermedad17–20.
El fenómeno de Lyonización descrito por la doctora Mery Lyon en 1961, es un mecanismo de compensación de la dosis genética, en el cual se silencia al azar, por diferentes mecanismos genéticos, un cromosoma X en células somáticas11–13.
El objetivo central de este estudio es describir el reporte de un caso clínico de EGC y la importancia del ensayo de Dihidrorodamina 1,2,3 por citometrìa de flujo, como apoyo en el diagnóstico de la EGC.
Materiales y métodosSe obtuvieron 5ml de muestra de sangre periférica en tubo con Heparina, 90ul de sangre fue incubada con PMA (1mg/ml) durante 20 minutos y dos tubos, cada uno con 90ul fueron dejados como control y uno solo incubado con PBS1x. Posteriormente la sangre periférica estimulada y una de las muestras no estimuladas fueron incubadas con 25ul de Dihidrorodamina 1,2,3 (1 ug/ml) durante 5 minutos. Los glóbulos rojos fueron posteriormente lisados con solución de amonio 0.168M durante 20 minutos. Finalmente se realizaron 2 lavados con PBS pH: 7.2.
La oxidación del colorante se analizó por citometrìa de flujo en un citometro FACS canto II de Becton Dickinson (BD), y se calcularon los valores de la intensidad media de fluorescencia (IMF) de neutrófilos del paciente, de la madre y de un control pareado por edad y género.
El índice oxidativo del neutrófilo (ION) fue calculado dividiendo la IMF del tubo estimulado sobre la intensidad media de florescencia del tubo con actividad basal.
Caso clinicoSe reporta el caso de dos pacientes; madre e hijo varón de 12 años de edad, producto de parto por cesárea prematuro de 34 semanas. Recibió maduración pulmonar, peso al nacer de 1800 gr. Antecedentes familiares: padres no consanguíneos, producto de sexta gestación, cuatro hermanos varones fallecidos por infecciones respiratorias y abdominales, así como la formación de granulomas antes del año de vida. Antecedente de dos sobrinos diagnosticados con EGC, hijos de una hermana portadora de la enfermedad. A continuación se muestra el Familiograma de acuerdo con la información citada anteriormente (fig. 1).
, III 1 padece enfermedad, III2 padece enfermedad.")
Familiograma. Se presenta I.1 sano I.2 portadora, II.1 sano, II.2portadora, II.3 padece enfermedad fallecida, II.4 padece enfermedad fallecido, II.5 padece enfermedad fallecido, II.6padece enfermedad fallecido, II7 (motivo de la publicación), III 1 padece enfermedad, III2 padece enfermedad.
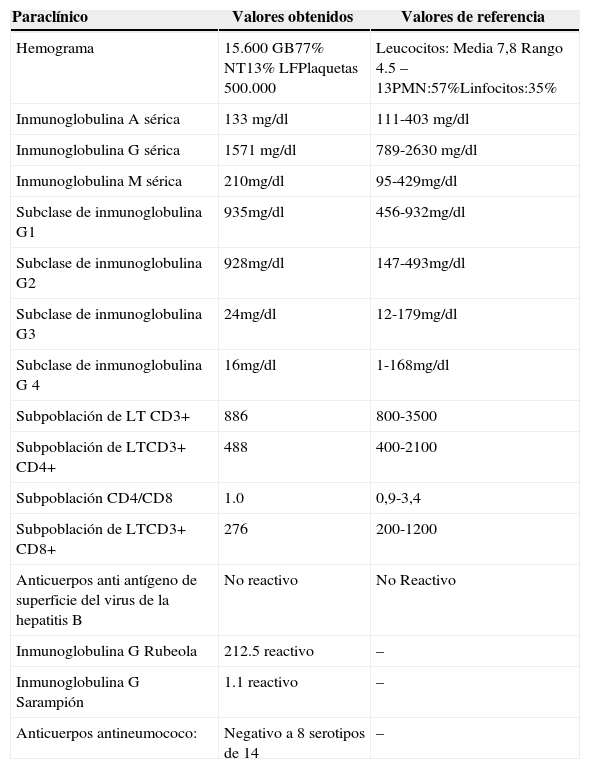
El paciente ha presentado múltiples cuadros infecciosos recurrentes desde los 7 meses de edad, que han requerido tratamiento hospitalario; cuatro neumonías, sinusitis crónica antes del año, infección por citomegalovirus y salmonelosis en dos ocasiones, celulitis periorbicular por gram positivos, no existen antecedentes de reacciones post vacunación. Las pruebas diagnósticas realizadas a los doce años de edad se presentan a continuación (tabla 1).
Paraclínicos realizados a los 12 años de edad
| Paraclínico | Valores obtenidos | Valores de referencia |
|---|---|---|
| Hemograma | 15.600 GB77% NT13% LFPlaquetas 500.000 | Leucocitos: Media 7,8 Rango 4.5 – 13PMN:57%Linfocitos:35% |
| Inmunoglobulina A sérica | 133 mg/dl | 111-403 mg/dl |
| Inmunoglobulina G sérica | 1571 mg/dl | 789-2630 mg/dl |
| Inmunoglobulina M sérica | 210mg/dl | 95-429mg/dl |
| Subclase de inmunoglobulina G1 | 935mg/dl | 456-932mg/dl |
| Subclase de inmunoglobulina G2 | 928mg/dl | 147-493mg/dl |
| Subclase de inmunoglobulina G3 | 24mg/dl | 12-179mg/dl |
| Subclase de inmunoglobulina G 4 | 16mg/dl | 1-168mg/dl |
| Subpoblación de LT CD3+ | 886 | 800-3500 |
| Subpoblación de LTCD3+ CD4+ | 488 | 400-2100 |
| Subpoblación CD4/CD8 | 1.0 | 0,9-3,4 |
| Subpoblación de LTCD3+ CD8+ | 276 | 200-1200 |
| Anticuerpos anti antígeno de superficie del virus de la hepatitis B | No reactivo | No Reactivo |
| Inmunoglobulina G Rubeola | 212.5 reactivo | – |
| Inmunoglobulina G Sarampión | 1.1 reactivo | – |
| Anticuerpos antineumococo: | Negativo a 8 serotipos de 14 | – |
Valores de referencia del hemograma tomados de Commans-Bitter WM, et al. J Pediatr 1997; 130(3):388-93.
Por el cuadro clínico presente en la paciente, además de los antecedentes personales y familiares, se realiza el test de Dihidrorodamina por citometrìa de flujo. La muestra de sangre periférica del paciente se procesó junto con la de su progenitora, además de un control pareado por edad y género. En el análisis de las muestras, se observó disminución severa de la producción de EROs en el paciente (ION:1,17) (fig. 2), con respecto al control (ION:94) (fig. 2), confirmándose en la madre el fenómeno de Lyonización, dada la presencia de dos poblaciones de Neutrófilos, una de ellas (Población N1: ION: 73,3) (fig. 2), con una producción de EROs normal, de acuerdo a lo observado en el control sano, y otra (Población N2: ION: 2,14) (fig. 2), con muy baja producción de EROs, similar a la detectada en el paciente. A continuación se muestran los histogramas obtenidos en cada uno de los casos14–22.
Discusión estimulados con (PMA) y sin estimulo (control sano (A), madre del paciente (B), paciente (C). En azul, actividad basal de la enzima y en rojo bajo el estímulo con PMA).")
La EGC en una inmunodeficiencia primaria en donde se presenta alteración funcional de los fagocitos, asociada a mutaciones en el complejo multiprotéico NADPH oxidasa, con un patrón de herencia predominante autosómico ligado al cromosoma X en el 80% de los casos, siendo los varones los más afectados, hijos de madres portadoras de la enfermedad.
Con respecto a los hallazgos clínicos del paciente, se puede concluir que las infecciones recurrentes sinopulmonares son la manifestación más frecuente, las cuales son de inicio temprano y por agentes infecciosos bacterianos. De igual manera, el número de infecciones es importante tener en cuenta en este caso, dado la presencia de más de un episodio de neumonía en un año, o de dos en dos años consecutivos, situación que es considerada como un signo de alerta en IDP. Por otra parte, es de vital importancia el diligenciamiento completo de la historia clínica, que incluya aspectos fundamentales como los antecedentes familiares de muertes tempranas por causas infecciosas de predominio en varones, además del número, etiología y severidad de cuadros clínicos infecciosos, así como la necesidad de tratamiento parenteral para el manejo de los mismos.
La presencia de respuesta inflamatoria granulomatosa, se asocia a múltiples casos de EGC con manifestaciones obstructivas en vía urinaria, piel, tejido pulmonar y tracto gastrointestinal. El antecedente de BCGitis se encuentra presente en el 40% de los casos, sin embargo, en este caso no hay registro de estas manifestaciones.
El uso de la prueba de oxidación de Dihidrorodamina 1, 2, 3 por citometrìa de flujo, permite realizar el diagnostico bioquímico de la enfermedad, además de una aproximación en el patrón de herencia. Según el estudio realizado por Jirapongsananuruk et al, 2003 la prueba de NBT presenta una menor sensibilidad, al ser una prueba cualitativa y no cuantitativa, operador dependiente. Con respecto al caso del presente artículo, la prueba de Dihidrorodamina 1, 2, 3 permite detectar una disminución severa en la producción de EROs, versus el control. Es importante afirmar que la prueba de NBT no fue realizada en este paciente18–20.
Con respecto a la madre del paciente, se detecta un padrón bimodal en el histograma, el cual se encuentra relacionado con el fenómeno de desviación genética o Lyonización, que se caracteriza por el silenciamiento o inactivación al azar de uno de los cromosomas X, presentándose desde el periodo embrionario gracias a la metilación del ADN y posteriormente por un ARN no traducible, situación que convierte a las madres en portadoras de la enfermedad. En este patrón bimodal, se puede concluir que se observan dos poblaciones de células, una de ellas con una producción de EROs comparable a la observada en el control (equivalente al 55%), y otra en la cual se observa disminución en la producción de EROs (equivalente 45%), resultados que son similares a los observados por Berron-Ruiz et al, 2014. La desviación genética extrema, se relaciona con madres sintomáticas portadoras de la enfermedad, sin embargo, de acuerdo con lo expresado anteriormente, en este reporte de caso no se presenta esta situación. En los pacientes con prueba de Dihidrorodamina positiva, con o sin confirmación molecular de esta enfermedad, es importante la respectiva asesoría genética18–20.
ConclusionesEn los pacientes con sospecha de inmunodeficiencia funcional de fagocitos, es fundamental el buen diligenciamiento de la historia clínica para correlacionar las diferentes manifestaciones clínicas, con el antecedente de infecciones severas frecuentes e inusuales, como con el antecedente familiar de muertes tempranas. La prueba de oxidación de Dihidrorodamina es fundamental frente a la sospecha clínica de EGC.
Los resultados observados en este estudio motivan la realización de pruebas moleculares confirmatorias del diagnóstico, además de la publicación de los valores del estallido respiratorio por grupo atareo observados en controles sanos.
Agradecimiento a la familia del paciente por permitir la construcción del conocimiento de la ciencia médica.