

Para obtener una autorización sanitaria de comercialización en la Unión Europea (UE), una compañía farmacéutica puede seguir uno de los dos procedimientos de registro europeos existentes en la actualidad: el procedimiento de registro centralizado y el procedimiento de registro de mutuo reconocimiento.
Procedimiento de registro centralizado
Es obligatorio en el caso de los medicamentos derivados de biotecnología, y se puede aplicar para otros medicamentos considerados muy innovadores. La autorización sanitaria de comercialización la concede la Agencia Europea de Evaluación de Medicamentos (EMEA) y es válida en todos los países de la Unión Europea1.
Procedimiento de registro de mutuo reconocimiento
Es útil para medicamentos convencionales cuando la compañía farmacéutica pretende comercializarlos en varios países europeos. A diferencia del procedimiento centralizado, la autorización sanitaria la concede cada una de las agencias nacionales de los diferentes países, que actúan reconociendo la primera autorización concedida por una de ellas.
Para medicamentos que se pretendan comercializar en un único Estado miembro de la Unión Europea la vía a seguir es el procedimiento de registro nacional2. En este tipo de procedimiento se solicita la autorización sanitaria al Estado miembro en el que se pretende comercializar el nuevo medicamento y la autorización concedida sólo es válida en ese Estado miembro. Este tipo de procedimiento es también el paso previo utilizado por una compañía farmacéutica para obtener la primera autorización e iniciar un procedimiento europeo de mutuo reconocimiento.
El procedimiento de registro de mutuo reconocimiento puede utilizarse para todo tipo de medicamentos excepto para aquellos obtenidos por alguno de los procesos biotecnológicos que
se relacionan en la parte A del anexo al Reglamento del Consejo 2.309/93/CEE
Normativa comunitaria y española
El procedimiento de mutuo reconocimiento se regula en la Directiva 93/39/CEE3 y el Reglamento 2.309/93/CEE4 del Consejo. En España, mediante el Real Decreto 2.000/955, se adecua la legislación española a la normativa comunitaria, y tiene carácter de legislación de productos farmacéuticos.
El procedimiento de registro de mutuo reconocimiento puede utilizarse para todo tipo de medicamentos excepto para aquellos obtenidos por alguno de los procesos biotecnológicos que se relacionan en la parte A del anexo al Reglamento del Consejo 2.309/93/CEE4, los cuales están obligados a seguir un procedimiento de registro centralizado y son los siguientes:
Tecnología del ADN recombinante.
Expresión controlada en los genes que codifican las proteínas biológicamente activas en procariotas y eucariotas, incluidas las células de mamífero transformadas.
Métodos basados en hibridomas y anticuerpos monoclonales.
Medicamentos veterinarios, incluidos los no obtenidos por biotecnología, empleados principalmente como potenciadores para fomentar el crecimiento o aumentar el rendimiento de los animales tratados.
El procedimiento de registro de mutuo reconocimiento se caracteriza porque una autorización de comercialización aprobada en un primer Estado miembro de la Unión Europea puede ser reconocida por las autoridades sanitarias competentes de otros Estados miembros sin que éstos tengan que volver a evaluar la calidad, seguridad y eficacia del medicamento. La unificación en los criterios normativos de evaluación en el conjunto de la Unión Europea hace posible este reconocimiento de unos estados con otros en la evaluación de los expedientes de registro.
En caso de que la compañía farmacéutica esté en desacuerdo con la decisión tomada por el Comité, tiene la posibilidad de iniciar un proceso de apelación ante el mismo, exponiendo las razones de desacuerdo dentro de los siguientes 60 días desde la toma de la decisión
Este procedimiento garantiza la calidad, la seguridad y la eficacia de un nuevo medicamento a la vez que posibilita un acceso más rápido de medicamentos nuevos al mercado comunitario.
En los casos de desacuerdo entre los diferentes Estados miembros para conceder la autorización sanitaria, por objeciones en la calidad, seguridad y/o eficacia del nuevo medicamento garantizada por el primer Estado miembro, se pone en funcionamiento el procedimiento de arbitraje. En este procedimiento interviene la Agencia Europea de Evaluación de Medicamentos (EMEA) para tomar una decisión única en los puntos conflictivos.
Etapas del procedimiento
El procedimiento de mutuo reconocimiento puede iniciarse cuando la compañía farmacéutica presenta el expediente de registro en paralelo en todos los Estados miembros de la Unión Europea donde pretende comercializar el nuevo medicamento. Uno de los Estados miembros evalúa el expediente de registro y toma una decisión, y el resto reconoce la decisión tomada por este primer estado. Esta situación puede plantearse de distintas maneras:
Uno de los Estados miembros donde la compañía farmacéutica ha presentado su expediente de registro decide iniciar activamente la evaluación e informa de ello a los demás Estados miembros. Estos Estados miembros pueden decidir suspender la evaluación para esperar a reconocer la decisión del primer estado.
Ningún Estado miembro se decide a iniciar la evaluación. En este caso, la elección del estado donde se llevará a cabo dicha evaluación debe ser por acuerdo entre todos los Estados miembros implicados en el procedimiento. Así, un Estado miembro evalúa el expediente de registro y los demás reconocen su decisión.
En un Estado miembro ya ha sido evaluado y autorizado con anterioridad el nuevo medicamento por procedimiento nacional. Ahora los demás Estados miembros piden al primero un informe de evaluación del medicamento y reconocen su decisión.
Desde la concesión de la autorización sanitaria de comercialización, es obligación de la compañía farmacéutica mantener el expediente de registro actualizado respecto a los datos en ella contenidos
Sin embargo, aunque es posible, éste no es el proceso más habitual en el procedimiento de mutuo reconocimiento.
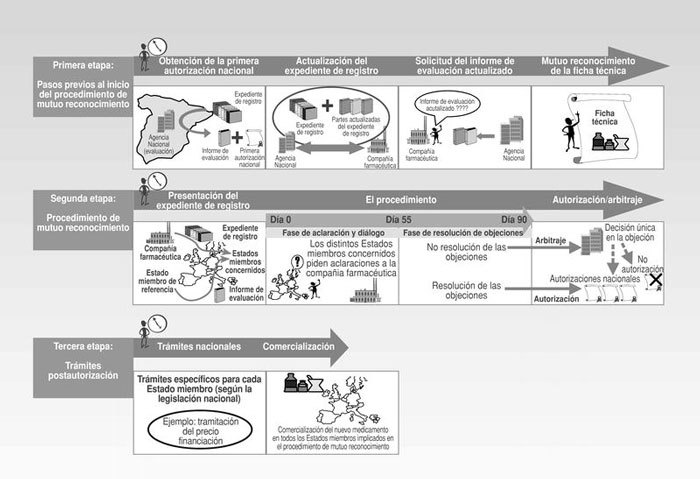
Normalmente, el procedimiento se inicia cuando una compañía farmacéutica elige un único Estado miembro donde solicita un procedimiento de registro nacional y obtiene la primera autorización que extenderá al resto de los Estados miembros europeos a través del procedimiento de mutuo reconocimiento. Este procedimiento desde la solicitud por parte de una compañía farmacéutica hasta la obtención de las correspondientes autorizaciones nacionales de comercialización, y, en caso de proceder, trámites específicos para cada país puede subdividirse en tres etapas (Fig. 1):
Pasos previos al inicio del procedimiento de mutuo reconocimiento.
Procedimiento de mutuo reconocimiento.
Trámites postautorización.
Pasos previos al inicio del procedimiento
La compañía farmacéutica debe presentar el expediente de registro del nuevo medicamento, a un único Estado miembro de la unión europea, y en este único Estado miembro debe iniciar un procedimiento de registro nacional. La autoridad sanitaria competente de este Estado miembro evalúa dicho expediente emite un informe de evaluación de la calidad, seguridad y eficacia del nuevo medicamento y concede la primera autorización sanitaria de comercialización (por ejemplo, en el caso de que la compañía farmacéutica decida escoger España como primer Estado de la UE donde iniciar un procedimiento de registro nacional, es la Agencia Española del Medicamento la autoridad sanitaria competente que evalúa el expediente de registro y emite un informe de evaluación en base al cual se concede la primera autorización sanitaria nacional de comercialización. Una vez obtenida esta primera autorización sanitaria nacional de comercialización, la compañía farmacéutica se encuentra en disposición de poder extender la autorización a más países de la Unión Europea a través del procedimiento de mutuo reconocimiento).
El país que concede la primera autorización se conoce como Estado miembro de referencia. Es preferible que una compañía farmacéutica informe desde el inicio al Estado miembro de referencia de su intención de utilizar esta primera autorización sanitaria nacional de comercialización para un posterior procedimiento de mutuo reconocimiento.
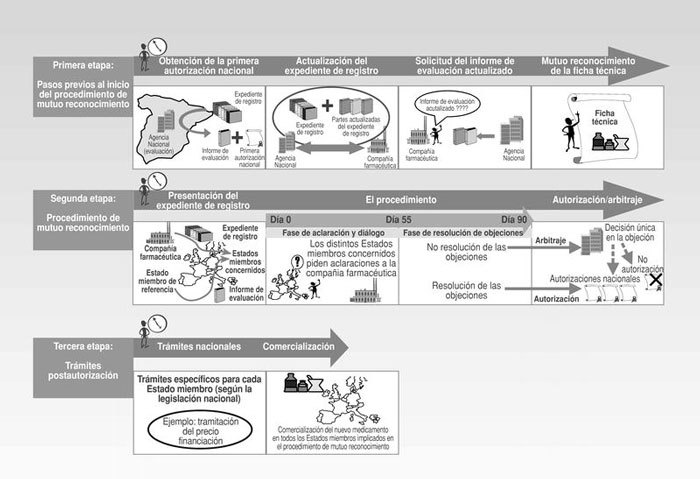
Fig. 1. Procedimiento de registro de mutuo reconocimiento.
Actualización del expediente de registro
Una vez obtenida la primera autorización sanitaria nacional de comercialización, la compañía decide cuándo desea iniciar el procedimiento europeo de mutuo reconocimiento, extendiendo la autorización a más Estados miembros de la Unión. En el tiempo transcurrido entre la autorización sanitaria nacional de comercialización y el inicio del procedimiento de mutuo reconocimiento, la compañía farmacéutica, debido al progreso científico y tecnológico, puede que introduzca cambios en la fabricación y control del medicamento autorizado. Por ello, antes de iniciar el procedimiento de mutuo reconocimiento la compañía farmacéutica y la autoridad sanitaria del Estado miembro de referencia se reúnen para discutir sobre los aspectos relevantes a actualizar del expediente de registro, asegurándose que toda la información será presentada en el procedimiento europeo.
Solicitud del informe de evaluación actualizado al Estado miembro de referencia
Como consecuencia de esta actualización del expediente de registro, la compañía farmacéutica solicita por escrito al Estado miembro de referencia un informe de evaluación actualizado, ya que este informe es la base para que la compañía farmacéutica pueda iniciar el procedimiento de registro de mutuo reconocimiento. En un plazo de 90 días desde la recepción de la solicitud, la autoridad sanitaria competente del Estado miembro de referencia debe poder proporcionar el informe de evaluación actualizado. En él deben constar las modificaciones que se hayan introducido desde la primera autorización sanitaria nacional de comercialización. Una vez preparado el informe, la autoridad sanitaria competente del Estado miembro de referencia lo notifica a la compañía farmacéutica.
Mutuo reconocimiento de la ficha técnica
Antes de iniciar el procedimiento de mutuo reconocimiento presentando el expediente de registro a otros Estados miembros de la Unión, el Estado miembro de referencia y la compañía farmacéutica deben prestar especial atención a la ficha técnica que se presentará del nuevo medicamento. La ficha técnica, según se define en el artículo 19 de la Ley 25/1990 del Medicamento, consiste en un resumen de la información científica esencial sobre la especialidad farmacéutica para ser difundida a los médicos y farmacéuticos y en ella deben constar los datos que figuran detalladamente en el anexo III del Real Decreto 767/93, de 21 de mayo6, como son el nombre, la composición y la forma farmacéutica del medicamento, los datos clínicos, las propiedades farmacológicas y los datos farmacéuticos. Su importancia reside en el hecho de que es la herramienta de trabajo de los profesionales sanitarios en el uso real del medicamento sobre la población.
En la redacción de la ficha técnica se tienen en cuenta las fichas técnicas existentes del mismo producto en los distintos Estados miembros y las fichas técnicas de productos con el mismo principio activo aprobados en previos procedimientos de mutuo reconocimiento. La manera de proceder en estas discusiones sobre la redacción de la ficha técnica entre el Estado miembro de referencia y la compañía farmacéutica siempre debe ser muy flexible.
Presentación del expediente de registro
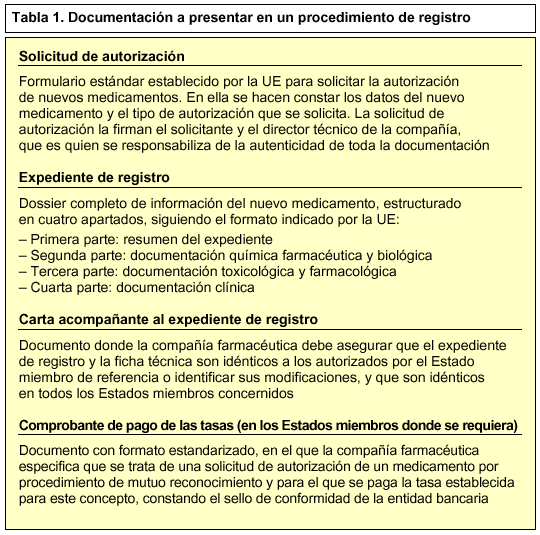
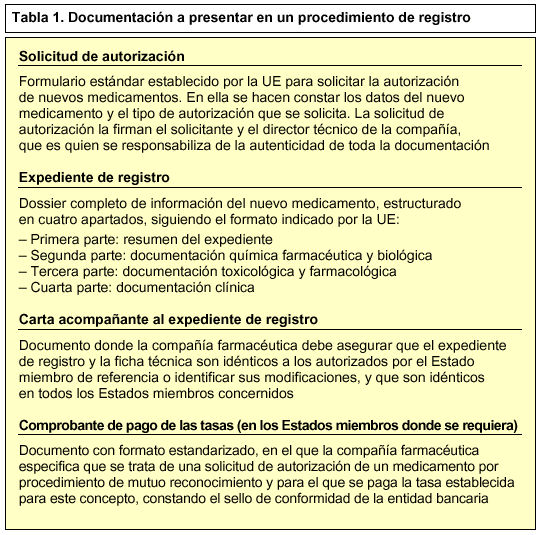
Para iniciar un procedimiento de mutuo reconocimiento, la compañía farmacéutica debe presentar a las autoridades sanitarias competentes de cada uno de los Estados miembros donde pretende comercializar el nuevo medicamento, la solicitud de autorización, el expediente de registro del nuevo medicamento, una carta acompañante y el comprobante de pago de tasas (tabla 1). Estos estados se conocen como Estados miembros concernidos. Además, enviará una notificación al Comité de Especialidades Farmacéuticas (CPMP) integrado en la EMEA.
La autoridad sanitaria competente del Estado miembro de referencia enviará una copia de su informe de evaluación a todos los Estados miembros concernidos y a la compañía farmacéutica.
El procedimiento
En cada uno de los Estados miembros concernidos se realiza la validación de la documentación, que consiste en una comprobación general de que se encuentra toda la documentación exigida. En caso de faltar alguna parte mínima de la documentación, se concede un plazo de 2 semanas para completarla.
El procedimiento de mutuo reconocimiento se inicia cuando:
La compañía farmacéutica informa tanto al Estado miembro de referencia como a los Estados miembros concernidos de la fecha de envío del expediente de registro a todos los Estados miembros concernidos,
El Estado miembro de referencia ha enviado el informe de evaluación a todos los Estados miembros concernidos y
No ha habido objeciones en el proceso de validación de la documentación.
El Estado miembro de referencia notifica que en 5 días empezará el procedimiento y que se extenderá durante 90 días. En estos 90 días, el Estado miembro de referencia actúa como punto central de unión entre la compañía farmacéutica y los Estados miembros concernidos.
Los primeros 55 días del procedimiento sirven para que los Estados miembros concernidos puedan pedir aclaraciones o realizar objeciones a determinados puntos del informe de evaluación que consideren importante resaltar. Estos puntos suelen estar mayoritariamente centrados en la ficha técnica propuesta por la compañía farmacéutica para el nuevo medicamento: indicaciones, posología y método de administración, contraindicaciones, advertencias especiales, precauciones en su uso, caducidad y condiciones de almacenamiento.
Entre el día 55 y el día 90 se intentan resolver todas las aclaraciones y objeciones planteadas por los diferentes Estados miembros concernidos mediante un diálogo con la compañía farmacéutica mediado por el Estado miembro de referencia. La compañía farmacéutica elabora un documento de respuesta estructurado en distintos apartados, según sean respuestas a la parte II (de calidad), a la parte III (de seguridad), a la parte IV (de eficacia) o a la ficha técnica. En este último aspecto, la compañía farmacéutica debe presentar una nueva propuesta de ficha técnica resaltando las modificaciones que realiza para responder a los distintos Estados miembros concernidos.
En esta fase de aclaraciones, objeciones y respuestas entre los Estados miembros y la compañía farmacéutica, cabe destacar el papel predominante de las llamadas break-out sessions (sesiones de exposición) dentro de las reuniones del Mutual Recognition Facilitation Group. Este último es un grupo creado en 1995 por las autoridades sanitarias competentes de los diferentes Estados miembros de la Unión Europea con el fin de coordinar y facilitar los procedimientos de mutuo reconocimiento. Está formado por un representante de cada Estado miembro y representantes de la Comisión Europea y de la EMEA. Se reúne una vez al mes en la EMEA, el lunes antes de las reuniones del CPMP. Paralelamente a estas reuniones, donde se tratan temas generales del procedimiento, se celebran las break-out sessions, encuentros coordinados por un Estado miembro de referencia en donde se solucionan puntos discordantes en la decisión de autorizar un nuevo medicamento en Europa por procedimiento de mutuo reconocimiento.
Después de esta fase, finalmente el Estado miembro de referencia y los Estados miembros concernidos llegan a un acuerdo. Normalmente, cada uno de los Estados miembros concernidos reconoce la autorización sanitaria de comercialización llegando a un acuerdo de ficha técnica en el plazo de los 90 días. Cada Estado miembro concernido que adopta la autorización y la ficha técnica informa de ello al Estado miembro de referencia, a los demás Estados miembros concernidos, al CPMP y a la compañía farmacéutica.
Arbitraje
En el caso excepcional de que, después de los 90 días de procedimiento de registro de mutuo reconocimiento, algún Estado miembro considere que existen serias razones para suponer que la concesión de la autorización sanitaria de comercialización va a desencadenar un riesgo para la salud pública (por la calidad, seguridad o eficacia del nuevo medicamento), se pone en funcionamiento el procedimiento de arbitraje. En este procedimiento de 90 días, el CPMP de la EMEA inicia una evaluación científica y toma una decisión única y vinculante en todos los Estados miembros en el punto conflictivo.
En caso de que la compañía farmacéutica esté en desacuerdo con la decisión tomada por el Comité, tiene la posibilidad de iniciar un proceso de apelación ante el mismo, exponiendo las razones de desacuerdo dentro de los siguientes 60 días desde la toma de la decisión. En 60 días más el CPMP decide si reconsiderar su posición y manifiesta su opinión definitiva. Esta decisión final es adoptada en 30 días por la Comisión Europea y por los Estados miembros que participan en el procedimiento.
Un expediente de registro puede ser retirado por la compañía farmacéutica en cualquier momento del procedimiento de mutuo reconocimiento y en cualquier Estado miembro, de manera que la compañía puede decidir continuar con el procedimiento en menos Estados miembros de los que había decidido al inicio, si observa objeciones que pudieran conducirle a un arbitraje. El arbitraje puede no interesar a una compañía farmacéutica ya que la opinión del CPMP, tanto si es positiva como negativa, es aplicable, sin excepción, a todos los Estados miembros implicados en el procedimiento.
En el caso de que la compañía farmacéutica retire el expediente de registro en uno o más Estados miembros concernidos durante el procedimiento de mutuo reconocimiento, no está permitido que presente luego en estos países un expediente para procedimiento de registro nacional.
Trámites postautorización
Una vez obtenida la autorización sanitaria de comercialización por el procedimiento de mutuo reconocimiento, el expediente de registro debe mantenerse armonizado en todos los Estados miembros.
Además, todas las nuevas formas farmacéuticas y dosificaciones que deriven de esta primera autorización también deberán seguir el procedimiento de mutuo reconocimiento.
En España, tras la obtención de la autorización sanitaria de comercialización quedan aún varios trámites hasta la puesta al mercado del nuevo medicamento al territorio nacional:
La AEM debe aprobar el embalaje y debe conceder el código nacional a la nueva especialidad farmacéutica.
La compañía farmacéutica debe negociar el precio con la Dirección General de Farmacia y Productos Sanitarios (DGFyPS).
Si procede, la compañía farmacéutica debe tramitar la financiación por el Sistema Nacional de la Salud (SNS).
Desde la concesión de la autorización sanitaria de comercialización, es obligación de la compañía farmacéutica mantener el expediente de registro actualizado respecto a los datos en ella contenidos. Toda modificación tecnológica en la fabricación y variaciones en los métodos de control de la especialidad farmacéutica deben ser de nuevo autorizados por las autoridades sanitarias competentes de cada uno de los Estados miembros de la UE. A su vez, las autorizaciones sanitarias tienen una validez temporal de 5 años. Transcurridos los 5 años, se debe revalidar la autorización.
La revalidación consiste en una evaluación del expediente de registro actualizado con las modificaciones que se hayan producido en el transcurso de los años, junto con datos referentes a farmacovigilancia para conseguir un mejor uso del medicamento. *