



Con la realización de este trabajo, las autoras intentan aproximar al conocimiento del lector la estructura de la Conferencia Internacional de Armonización (ICH) en las fases del procedimiento de armonización. Asimismo, presentan y comentan el denominado Documento Técnico Común (CTD, según las siglas en inglés) para el registro de medicamentos de uso humano.

En su origen, el registro de medicamentos suponía la obligación de que todos los nuevos medicamentos tuvieran que pasar por una fase de evaluación previa a su comercialización por parte de las autoridades sanitarias de sus propios países. Desde esta situación inicial de competencia nacional de cada país, el registro de medicamentos ha evolucionando hasta permitir la racionalización y armonización de los requerimientos exigidos entre los distintos países para determinar la seguridad, la eficacia y la calidad de un mismo medicamento.
Esta necesidad de racionalizar y armonizar las regulaciones nacionales estuvo encabezada por Europa en 1980 cuando se inició el desarrollo de un mercado único para productos farmacéuticos. Un poco más tarde se estableció el diálogo entre Europa, Estados Unidos y Japón sobre la posibilidad de armonización de requerimientos en las tres regiones. En 1990, fruto de este diálogo, nacieron las Conferencias Internacionales de Armonización de requerimientos técnicos para el registro de medicamentos de uso humano (ICH del inglés International Conference on Harmonization).
La ICH consiste en un proyecto conjunto de las autoridades reguladoras y la industria farmacéutica procedente de Europa, Estados Unidos y Japón, regiones donde se desarrolla la mayoría de nuevos medicamentos
ICH
La ICH consiste en un proyecto conjunto de las autoridades reguladoras y la industria farmacéutica procedente de Europa, Estados Unidos y Japón, regiones donde se desarrolla la mayoría de nuevos medicamentos, unidos en el objetivo de armonizar los requisitos técnicos y científicos exigidos para el registro de un medicamento. Esta armonización, a escala casi mundial, pretende conseguir un doble beneficio: la racionalización en el uso de recursos animales, humanos y también materiales para el desarrollo de nuevos medicamentos y la reducción del tiempo de disposición de un nuevo medicamento en el mercado.
La ICH consigue un acercamiento de las tres regiones haciendo posible la identificación de temas donde existen divergencias que hay que armonizar y el establecimiento de un método de trabajo común para la elaboración de recomendaciones o guías comunes para la industria farmacéutica y las autoridades reguladoras.
Los temas escogidos para la armonización se engloban dentro de tres categorías que corresponden a los criterios basándose en los cuales se autoriza un nuevo medicamento: calidad, seguridad y eficacia. Actualmente existen once temas de armonización distribuidos entre cada una de las tres categorías.
Las conferencias internacionales de armonización se celebran aproximadamente cada dos años. La primera tuvo lugar en 1991, y desde entonces se han celebrado cinco conferencias. Las conferencias se celebran alternando entre las tres regiones ICH por este orden (Europa, Estados Unidos, Japón) son las siguientes:
ICH 1. Bruselas, Bélgica en 1991.
ICH 2. Orlando, EE.UU. en 1993.
ICH 3. Yokohama, Japón en 1995.
ICH 4. Bruselas, Bélgica en 1997.
ICH 5. San Diego, EE.UU. en 2000.
La próxima conferencia se celebrará el próximo mes de noviembre en Osaka, Japón (ICH 6). Estas conferencias abren el foro de armonización a toda la comunidad científica, exponiéndose qué temas de regulación de medicamentos se han discutido y qué logros se han alcanzado.
Estructura de la ICH
En la ICH participan como patrocinadores seis grupos procedentes tanto de las autoridades reguladoras como de la industria farmacéutica de Europa, Estados Unidos y Japón. Por parte de las autoridades reguladoras participan:
La Comisión Europea.
El Ministry of Health, Labor and Welfare (MHLW) de Japón.
La Food and Drug Administration (FDA) de Estados Unidos.
Por parte de la industria farmacéutica participan:
La European Federation of Pharmaceutical Industries and Associations (EFPIA) de Europa.
La Japan Pharmaceutical Manufacturers Association (JPMA) de Japón.
La Pharmaceutical Research and Manufacturers of America (PhRMA) de Estados Unidos.
A continuación pasamos a comentar brevemente las características de cada uno de estos organismos.
Comisión Europea
La Comisión Europea participa en representación de los quince estados miembros. Hay que destacar que la Comisión Europea también trabaja, por su parte, en la armonización de los requerimientos técnicos y científicos exigidos para el registro de un medicamento en los distintos Estados miembros y conseguir un mercado único, elaborando también sus recomendaciones o guías.
MHLW
El Ministerio de Sanidad Japonés (MHLW) junto al Instituto Nacional de Ciencias para la Salud (National Insitute of Health Sciencies, NIHS, miembro afiliado al MHLW) son los organismos que participan directamente con la ICH.
FDA
La FDA es la autoridad responsable de la aprobación de todos los medicamentos utilizados en Estados Unidos. Se organiza en distintos centros responsables de los distintos medicamentos que se regulan y en concreto, en la ICH, participa el Center for Drug Evaluation and Research (CDER) y el Center for Biologics Evaluation and Research (CBER).
EFPIA
Participa en representación de asociaciones nacionales de la industria farmacéutica, por ejemplo, la Asociación Nacional Empresarial de la Industria Farmacéutica (Farmaindustria) es miembro de EFPIA y compañías farmacéuticas dedicadas a la investigación, el desarrollo y la fabricación de medicamentos de uso humano en Europa.
En total, EFPIA representa el punto de vista de más de 2.000 compañías farmacéuticas dedicadas a la investigación, el desarrollo y la fabricación de medicamentos de uso humano en Europa.
JPMA
La JPMA participa en representación de 90 compañías farmacéuticas dedicadas a la investigación de medicamentos.
PhRMA
Participa en representación de la industria investigadora en Estados Unidos. La asociación consta de 67 compañías dedicadas al descubrimiento, desarrollo y fabricación de medicamentos.
Además de la participación directa de estos seis grupos, cabe destacar la presencia de observadores que actúan de unión entre la ICH y los países o regiones de una área no-ICH. Entre estos observadores está la Organización Mundial de la Salud (OMS) y las autoridades reguladoras de Canadá y Suiza. Su presencia es importante para que los beneficios obtenidos puedan ser utilizados a escala mundial. También participa la International Federation of Pharmaceutical Manufacturers Association (IFPMA) en representación de la industria de 56 países en el mundo.
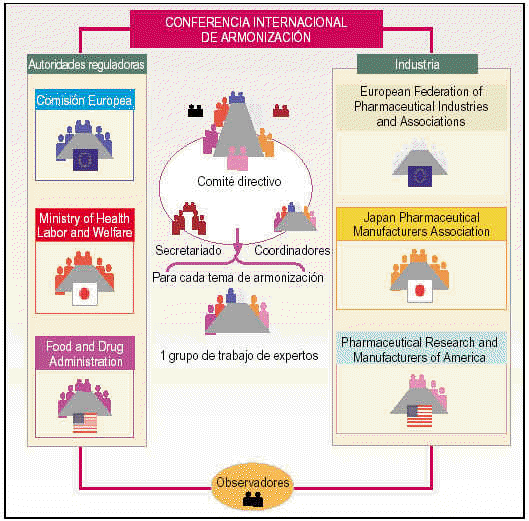
Todos estos participantes constituyen la estructura de la ICH que se organiza en (fig. 1):
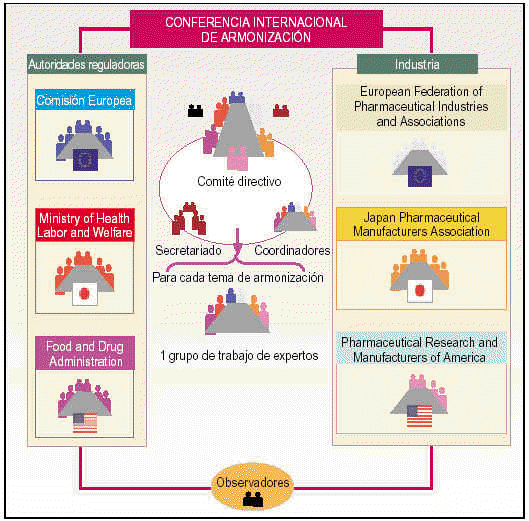
Fig. 1. Esquema de la ICH.
Comité directivo.
Coordinadores.
Secretariado.
Grupos de trabajo formados por expertos.
Comité directivo
Está formado por un total de 12 miembros (dos miembros de cada uno de los seis grupos patrocinadores). Sus principales funciones son las de determinar la política y los procedimientos de la ICH, seleccionar los temas de armonización y monitorizar el seguimiento de las iniciativas ICH.
El comité directivo se reúne como mínimo dos veces al año. Los observadores e IFPMA también están presentes en las reuniones del comité directivo aunque no tienen derecho a voto.
Coordinadores
Los forman un miembro de cada uno de los seis grupos ICH. La función del coordinador es la de asegurar que los documentos son distribuidos de forma correcta y a las personas adecuadas mediante el contacto directo con el secretariado.
Secretariado
Es responsable de la preparación de las reuniones del comité directivo y su documentación, de la coordinación de las reuniones de los grupos de trabajo y la preparación de la documentación técnica de las conferencias y de la relación con los conferenciantes.
Grupos de trabajo
Para cada tema de armonización seleccionado, el comité directivo designa un grupo de trabajo (Expert Working Group, EWG) para revisar las diferencias existentes entre Europa, Estados Unidos y Japón y alcanzar un consenso científico. Los grupos de trabajo están representados por los 6 grupos patrocinadores (UE, EFPIA, MHW, JPMA, FDA y PhRMA) y se reúnen la misma semana que el comité directivo para informarle sobre los progresos y objetivos alcanzados.
Cada unos de los seis grupos ICH también establecen una red de expertos para tratar el mantenimiento de recomendaciones ya existentes.
Fases del proceso de armonización
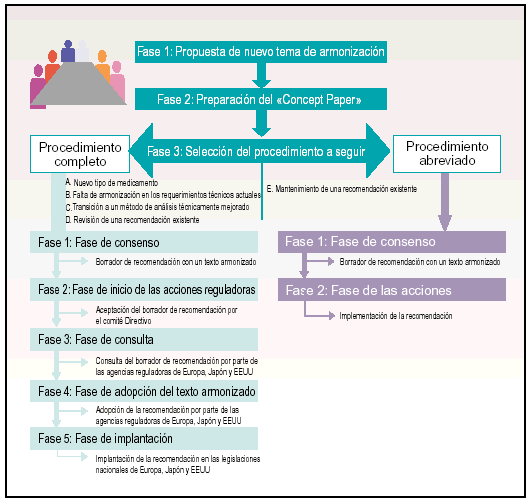
El proceso de armonización se puede agrupar en tres fases principales (fig. 2):
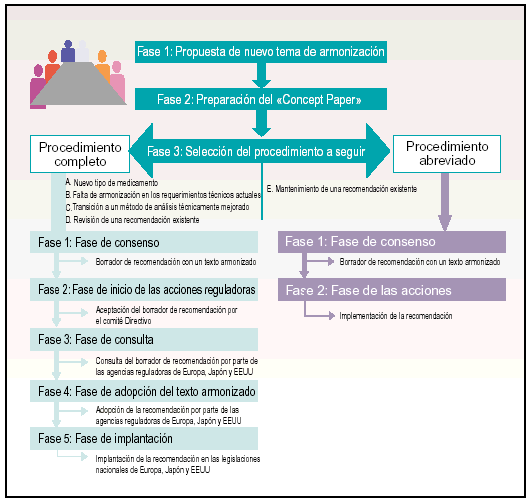
Fig. 2. Fases del proceso de armonización.
Fase 1. Propuesta de un nuevo tema de armonización.
Fase 2. Preparación del Concept Paper.
Fase 3. Selección del procedimiento a seguir: procedimiento completo o procedimiento abreviado.
Propuesta de una nueva iniciativa de armonización
Un nuevo tema de armonización puede surgir de ámbitos muy distintos (asociaciones, federaciones o sociedades reconocidas de profesionales otras conferencias regionales o internacionales, simposios), pero cualquiera que sea la fuente, la propuesta formal a la ICH debe canalizarse a través de uno de los 6 grupos patrocinadores o de uno de los observadores.
La nueva propuesta de armonización realizada puede pertenecer a distintas categorías:
Nuevo tipo de medicamento. Es una recomendación para cubrir nuevos medicamentos resultantes del avance de la tecnología y las técnicas para desarrollar medicamentos. Por ejemplo, una recomendación para medicamentos surgidos de la terapia génica.
Cada unos de los seis grupos ICH también establecen una red de expertos para tratar el mantenimiento de recomendaciones ya existentes
Falta de armonización en los requerimientos técnicos actuales. Es una recomendación para incrementar la armonización en una recomendación ya existente.
Transición a un método de análisis técnicamente mejorado.
Es una recomendación para facilitar la sustitución de un método establecido por un método de análisis mejor.
Revisión de una recomendación existente. Es una recomendación para introducir cambios significativos a los aspectos técnicos de una recomendación ya existente.
Mantenimiento de una recomendación existente. Es una recomendación para introducir pequeños cambios, actualizaciones, revisiones para tener en cuenta problemas surgidos en la implantación o nueva información de una recomendación ya existente.
Preparación del Concept Paper
El grupo patrocinador o el observador que va a realizar la propuesta de la nueva iniciativa de armonización debe presentar ante el comité directivo un Concept Paper. El Concept Paper es un documento de no más de dos páginas de extensión, en el que se resume la propuesta en forma de cinco apartados:
Apartado 1. Tipo de acción armonizadora que se propone. En este apartado se describe la propuesta de acción armonizadora de acuerdo a la categoría que corresponda: nuevo tipo de medicamento, falta de armonización en los requerimientos técnicos actuales, transición a un método de análisis técnicamente mejorado, revisión de una recomendación existente y mantenimiento de una recomendación existente.
Apartado 2. Declaración de problema percibido. En este apartado se indica brevemente los problemas que está o que va a causar la falta de armonización.
Apartado 3. Cuestiones a resolver. En este apartado se listan resumidamente los principales puntos científicos y técnicos que requieren armonización.
Apartado 4. Antecedentes de la propuesta. En este apartado se incluye otra información que se considere relevante como puede ser el origen de la propuesta o referencias a publicaciones.
Apartado 5. Tipo de grupo de experto. En este apartado se realiza una propuesta del tipo de grupo de experto que se requeriría para el tema de armonización que se propone. Hay distintas opciones, tal como muestra la tabla 1.

Selección del procedimiento a seguir: procedimiento completo o procedimiento abreviado
Existen dos tipos de procedimientos a seguir según la categoría a la que corresponda la nueva propuesta de armonización. El procedimiento completo se aplica a las siguientes categorías:
Nuevo tipo de medicamento.
Falta de armonización en los requerimientos técnicos actuales.
Transición a un método de análisis técnicamente mejorado.
Revisión de una recomendación existente.
Por su parte, el procedimiento abreviado se aplica a la categoría de mantenimiento de una recomendación existente.
En el procedimiento completo el Concept Paper se envía a los coordinadores con el fin de que los distintos grupos ICH puedan realizar sus comentarios. Una vez recibidos los comentarios el Concept Paper, junto a estos comentarios, se envía al comité directivo quien determina si la propuesta es de suficiente interés para todos los grupos como para incorporarla al programa de trabajo de la ICH o es preferible desestimarla.
Si el comité directivo decide aceptar la propuesta, revisará el Concept Paper para incorporar los siguientes aspectos:
Los objetivos y los resultados esperados de la propuesta armonizadora.
La composición del EWG.
El calendario y el plan de acción del EWG, que no suele sobrepasar los 2 años.
Los observadores también podrán designar un experto para el grupo de trabajo.
Fases ulteriores de armonización
A partir de este momento, el procedimiento que se sigue para alcanzar la armonización en cada uno de los temas que se proponen, se subdivide en cinco fases más:
Fase 1. Fase de consenso.
Fase 2. Fase de inicio de las acciones reguladoras.
Fase 3. Fase de consulta.
Fase 4. Fase de adopción del texto armonizado.
Fase 5. Fase de implantación.
En el procedimiento completo el Concept Paper se envía a los coordinadores con el fin de que los distintos grupos ICH puedan realizar sus comentarios
Fase de consenso
El ponente, representado por un miembro del grupo de trabajo, actúa como coordinador de la fase de consenso. En esta fase el objetivo es conseguir un acuerdo por parte del grupo de trabajo en representación de los seis grupos patrocinadores. Para ello, el grupo de trabajo intercambia sus opiniones hasta alcanzar un borrador de recomendación con un texto consensuado.
Transcurrido el tiempo fijado en el calendario del Concept Paper puede que se haya llegado a un texto consensuado, que se presenta ante el comité directivo, o que no se haya podido llegar a un texto consensuado. En este caso, se debe presentar un informe al comité directivo indicando los puntos de acuerdo y señalando los puntos en los que existe divergencia entre las partes implicadas. En este momento el comité directivo puede decidir entre:
Permitir una extensión del calendario si observa que el consenso puede alcanzarse en un período corto que se especificará.
Suspender o abandonar el proyecto de propuesta armonizadora.
Pasar a la siguiente fase con el texto a pesar de que no exista un consenso total.
Fase de inicio de las acciones reguladoras
En esta fase el comité directivo acepta el borrador de recomendación consensuado por el grupo de trabajo.
Fase de consulta
En esta fase, el documento abandona el grupo de trabajo y se envía a las agencias reguladoras de las tres regiones ICH (Europa, Estados Unidos y Japón) para consulta de acuerdo con los procedimientos habituales de cada institución:
En la Unión Europea, el texto se presenta ante el Comité de especialidades Farmacéuticas (CPMP), quien decide la duración de la fase de consulta con las partes interesadas (habitualmente 6 meses). La recomendación se publica y distribuye por la EMEA como borrador de directriz del CPMP.
En Estado Unidos, el texto se publica por la Food and Drug Administration como borrador de guía en el Registro Federal con una fecha límite de recepción de comentarios por escrito.
En Japón, el texto se traduce al japonés y se notifica para comentarios por el Pharmaceutical and Medical Safety Bureau (PMSB) integrado dentro del Ministry of Health, Labor and Welfare (MHLW), quien decide la duración de la fase de consulta.
Los comentarios recibidos se analizan por parte de las tres autoridades reguladoras representadas en el grupo de trabajo (UE, FDA, MHW) y se va modificando la recomendación, tantas veces como sea necesario, hasta llegar a un consenso. En esta etapa, por tanto, el coordinador --denominado «ponente regulatorio»-- será uno de entre los tres miembros que representan las autoridades reguladoras en el grupo de trabajo. Una vez alcanzado el consenso, el borrador es enviado por el ponente regulatorio al comité directivo para su adopción.
Fase de adopción del texto armonizado
El comité directivo recibe el documento del ponente regulatorio y lo recomienda a las autoridades reguladoras de las tres regiones ICH para su adopción.
En el caso en que alguno de los grupos representantes de la industria tenga objeciones en la adopción de la recomendación, los grupos representantes de las autoridades regulatorias pueden decidir que el texto revisado se libere de nuevo a consulta.
Fase de implantación
En esta última fase, el texto armonizado se incorpora a las legislaciones nacionales de acuerdo al procedimiento habitual de cada región:
En Europa, el CPMP establece el tiempo de implantación (habitualmente 6 meses). Transcurrido este tiempo, la recomendación es publicada por la Comisión Europea en el volumen 3 de las normas que rigen los medicamentos de uso humano en la Unión Europea (Rules Governing Medicianl Products in the European Union).
En Estados Unidos, la FDA publica el texto de la recomendación en el Federal Register. El día de publicación en el Federal Register es el día en que la recomendación se considera implantada para su utilización.
En Japón, la recomendación traducida al japonés es notificada por el Pharmaceutical and Medical Safety Bureu con una fecha de implantación.
En el procedimiento abreviado el Concept Paper se envía al secretariado y a los coordinadores para que se pueda iniciar el proceso de mantenimiento. Los coordinadores son quienes determinan si la propuesta constituye un cambio menor y, por tanto, debe iniciarse el proceso de mantenimiento.
Fases del mantenimiento de la recomendación
El procedimiento que se sigue para alcanzar el mantenimiento de la recomendación se subdivide en dos fases:
Fase 1. Fase de consenso.
Fase 2. Fase de las acciones.
Fase de consenso
En esta fase el objetivo es conseguir un acuerdo por parte del grupo de trabajo de mantenimiento en representación de los seis grupos patrocinadores. Para ello el grupo de trabajo intercambia sus opiniones hasta alcanzar un borrador de recomendación con un texto consensuado.
Puede que se haya llegado a un texto consensuado que se presenta ante el comité directivo. Si no se ha llegado a un consenso, la propuesta se remite a la próxima reunión del comité directivo para que éste decida si establecer un grupo de expertos e iniciar un procedimiento completo
Fase de las acciones
En esta fase la propuesta es implementada de la siguiente manera:
Sin consulta, mediante el anuncio y la publicación del texto revisado.
Previa consulta por parte de las autoridades reguladoras. La propuesta se trata como la fase 2 del procedimiento completo anteriormente explicado.
El trabajo de la ICH
Como ya se ha comentado anteriormente, los temas de armonización están englobados dentro de tres categorías que corresponden a los criterios a partir de los cuales se aprueba y autoriza un nuevo medicamento: calidad, seguridad y eficacia. Hay una cuarta categoría multidisciplinaria, para temas de armonización que no pueden ser englobados específicamente en una de las tres categorías anteriores. Los temas de armonización que van surgiendo de la ICH se codifican atendiendo a la categoría a la que corresponden:
Los temas de armonización relacionados con la calidad del medicamento son codificados con la letra Q (del inglés quality) seguida de un número correlativo. En esta categoría encontraremos temas sobre aspectos químicos, farmacéuticos y biológicos del medicamento. Actualmente existen 7 temas de armonización asignados bajo la categoría Q de calidad (de Q1 a Q7) con 19 recomendaciones.
Los temas de armonización relacionados con la seguridad del medicamento son codificados con la letra S (del inglés safety) seguida de un número correlativo. En esta categoría encontraremos temas sobre estudios preclínicos in vitro e in vivo. Actualmente existen 7 temas de armonización asignados bajo la categoría S de seguridad (de S1 a S7) con 13 recomendaciones.
Los temas de armonización relacionados con la eficacia del medicamento son codificados con la letra E (del inglés efficacy) seguida de un número correlativo. En esta categoría encontraremos temas sobre estudios clínicos en humanos. Actualmente existen 12 temas de armonización asignados bajo la categoría E de eficacia (de E1 a E12) con 14 recomendaciones.
Los temas de armonización no relacionados exclusivamente con la calidad, la seguridad y la eficacia del medicamento son codificados con la letra M (del inglés multidisciplinary) seguida de un número correlativo. Actualmente existen 4 temas de armonización asignados bajo la categoría M de multidisciplinar (de M1 a M4) con 4 recomendaciones.
CTD
El objetivo de la primera fase de las actividades de la ICH fue el de armonizar el contenido técnico de aquellas secciones donde existían mayores divergencias entre los requerimientos exigidos por las autoridades reguladoras de las tres regiones ICH, con el objetivo de evitar redundancias y duplicidades en los procedimientos de desarrollo de los medicamentos.
Alcanzada esta primera etapa en la que un grupo común de datos se genera para demostrar la calidad, seguridad y eficacia de un nuevo medicamento en cualquiera de las tres regiones ICH, pareció el momento idóneo para ver la viabilidad de desarrollar un documento técnico común para reportar estos datos a las autoridades reguladoras.
El CTD pretende ser un dossier de registro común y válido a escala mundial. Este dossier común garantizaría la calidad, seguridad y eficacia del medicamento y sería aceptado en las agencias evaluadoras de casi todo el mundo, lo que supondría un gran beneficio tanto para las compañías farmacéuticas como para la población mundial.
El CTD está codificado como tema M4. Documento Técnico Común para el registro de medicamentos de uso humano, con tres recomendaciones:
Se debe continuar con la selección de nuevos temas de armonización, que muy probablemente procederán de los últimos avances tecnológicos en medicamentos y de actividades poscomercialización
M4Q. Common Technical Document for the registration of pharmaceuticals for human use (quality).
M4S. Common Technical Document for the registration of pharmaceuticals for human use (safety).
M4E. Common Technical Document for the registration of pharmaceuticals for human use (efficacy).
Estas recomendaciones alcanzaron la fase 2 de aprobación por parte del comité directivo en julio de 2000. En septiembre de 2000 iniciaron la fase 3 de consulta y fueron enviadas a las agencias reguladoras de las tres regiones ICH (Europa, Estados Unidos y Japón) para que pudieran realizar sus comentarios hasta la fecha de septiembre de 2000. El 9 de noviembre de 2000 se encontraban en la fase 4 en la que el comité directivo recomendó su adopción a las autoridades reguladoras de las tres regiones ICH. La versión de noviembre de 2000 del CTD ha superado la fase 5 de implantación en las tres regiones ICH, aunque existe una revisión de septiembre de 2002 pendiente de implantación.
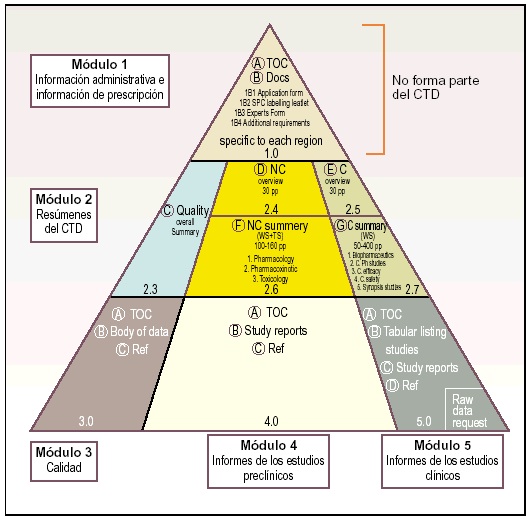
El CTD se estructura en 5 módulos, de los cuales el módulo 1 es específico de cada una de las regiones ICH y los módulos 2, 3, 4 y 5 son propiamente el formato común:
Módulo 1. Información administrativa e información de prescripción.
Módulo 2. Resúmenes del CTD.
Módulo 3. Calidad.
Módulo 4. Informes sobre los estudios preclínicos.
Módulo 5. Informes sobre los estudios clínicos.
Estos cinco módulos que componen el CTD se representan gráficamente a través de una pirámide (fig. 3) en la que los módulos 3, 4 y 5 que avalan respectivamente la calidad, seguridad y eficacia del medicamento se encuentran en la base, el módulo 2 con los resúmenes y revisiones en el centro y la información regional en la punta.
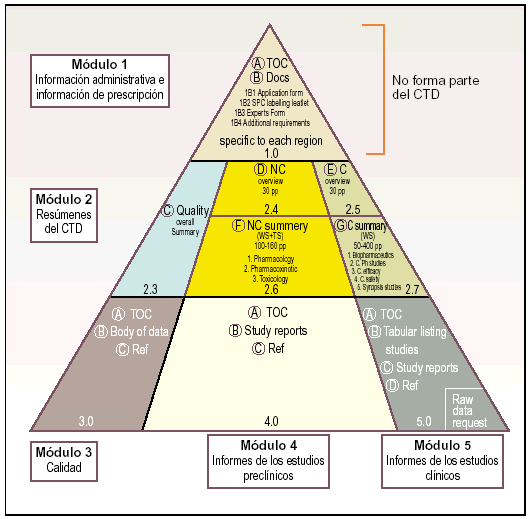
Fig. 3. Esquema piramidal de los módulos del CTD.
El futuro de la ICH
Después de más de 10 años del inicio de las actividades de la ICH, el futuro es continuar con sus actividades de armonización. Después de haber conseguido la armonización de criterios técnicos y finalmente la armonización de las solicitudes de registro, es importante en el futuro mantener estos logros y centrarse en su implementación y monitorización. Además se debe continuar con la selección de nuevos temas de armonización, que muy probablemente procederán de los últimos avances tecnológicos en medicamentos y de actividades poscomercialización.