



Revisión legislativa del derecho farmacéutico comunitario
Hasta el 30 de abril de 2004, el panorama legislativo comunitario en materia de registro de medicamentos de uso humano estaba regulado en dos normas: en la Directiva 2001/83/CE del Parlamento Europeo y del Consejo y en el Reglamento 2.309/93 del Consejo. Desde entonces, fruto de un intenso proceso legislativo, se aprobaron las disposiciones que actualmente configuran la estructura legislativa del derecho farmacéutico comunitario. Las autoras en este trabajo analizan la Directiva 2004/27 por la que se modifica el código comunitario sobre medicamentos de uso humano.

El Parlamento Europeo y el Consejo aprobaron en noviembre de 2001 la Directiva 2001/83/CE, mediante la que se establece un código comunitario sobre medicamentos de uso humano. Con la publicación del código, el panorama legislativo comunitario en materia de registro de medicamentos de uso humano se regula mediante la citada directiva y el Reglamento 2.309/93 del Consejo, de 22 de julio.
A pesar del buen funcionamiento del sistema de regulación de medicamentos, el propio Reglamento 2.309/93, en concreto su artículo 71, exigía que en un plazo de 6 años a partir de su entrada en vigor (es decir, hasta 2001), la Comisión Europea presentaría un informe general sobre la experiencia adquirida como resultado de la aplicación de los procedimientos comunitarios. En cumplimiento de lo establecido, la Comisión Europea publicó en noviembre de 2001 el citado informe exponiendo las conclusiones de la evaluación del funcionamiento de los procedimientos de registro y sus propuestas de revisión. Desde entonces ha habido un intenso proceso legislativo a través de la Comisión, el Parlamento y el Consejo Europeo hasta la adopción de los documentos que configuran hoy la revisión legislativa del derecho farmacéutico comunitario:
* La Directiva 2004/27/CE del Parlamento Europeo y del Consejo de 31 de marzo de 2004 que modifica la Directiva 2001/83/CE por la que se establece un código comunitario sobre medicamentos de uso humano (DO L 136, 30.4.2004).
* La Directiva 2004/24/CE del Parlamento Europeo y del Consejo de 31 de marzo de 2004 por la que se modifica, en lo que se refiere a los medicamentos tradicionales a base de plantas, la Directiva 2001/83/CE por la que se establece un código comunitario sobre medicamentos para uso humano (DO L 136, 30.04.2004).
* El Reglamento 726/2004 del Parlamento Europeo y del Consejo de 31 de marzo de 2004 por el que se establecen procedimientos comunitarios para la autorización y el control de los medicamentos de uso humano y veterinario, y por el que se crea la Agencia Europea de Medicamentos (DO L 136, 30.04.2004).
En concreto, la Directiva 2004/27/CE responde a cuatro objetivos básicos:
* Incrementar la salud del paciente europeo mediante una mayor vigilancia del mercado europeo de los medicamentos.
* Asegurar que el sistema legislativo europeo es viable frente a la ampliación de la Unión Europea.
* Racionalizar y simplificar el sistema, para hacerlo más coherente y transparente.
* Favorecer la competitividad de la industria farmacéutica.
Los puntos más destacables de la reforma impuesta por la Directiva 2004/27/CE se centran en los siguientes aspectos:
* Definiciones y ámbito de aplicación.
* Autorización de comercialización y procedimiento de registro.
* Procedimiento de Registro de Reconocimiento Mutuo.
* Medicamentos genéricos.
* Inspección y control e impacto medioambiental.
* Farmacovigilancia.
* Información proporcionada a los pacientes.
Definiciones y ámbito de aplicación
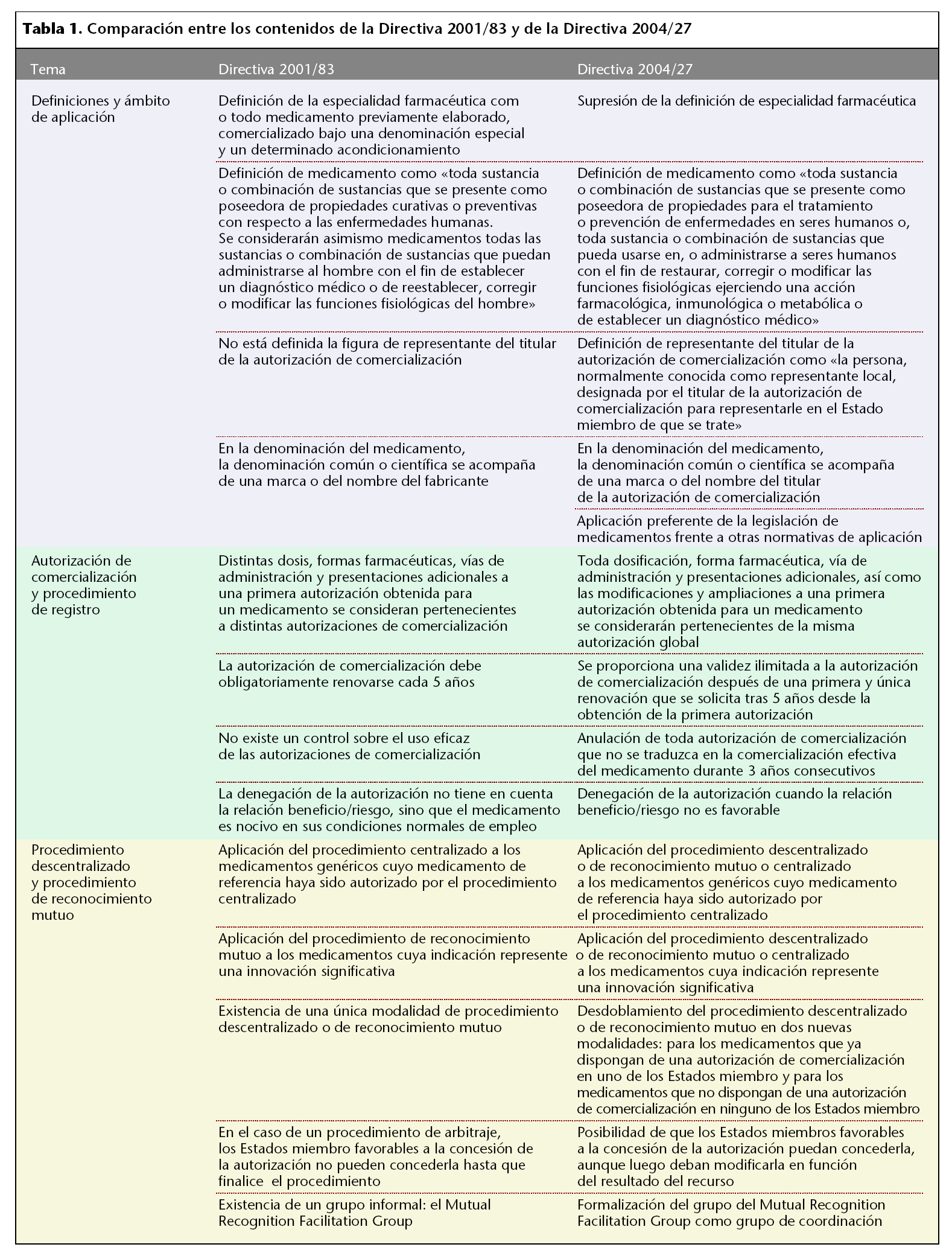
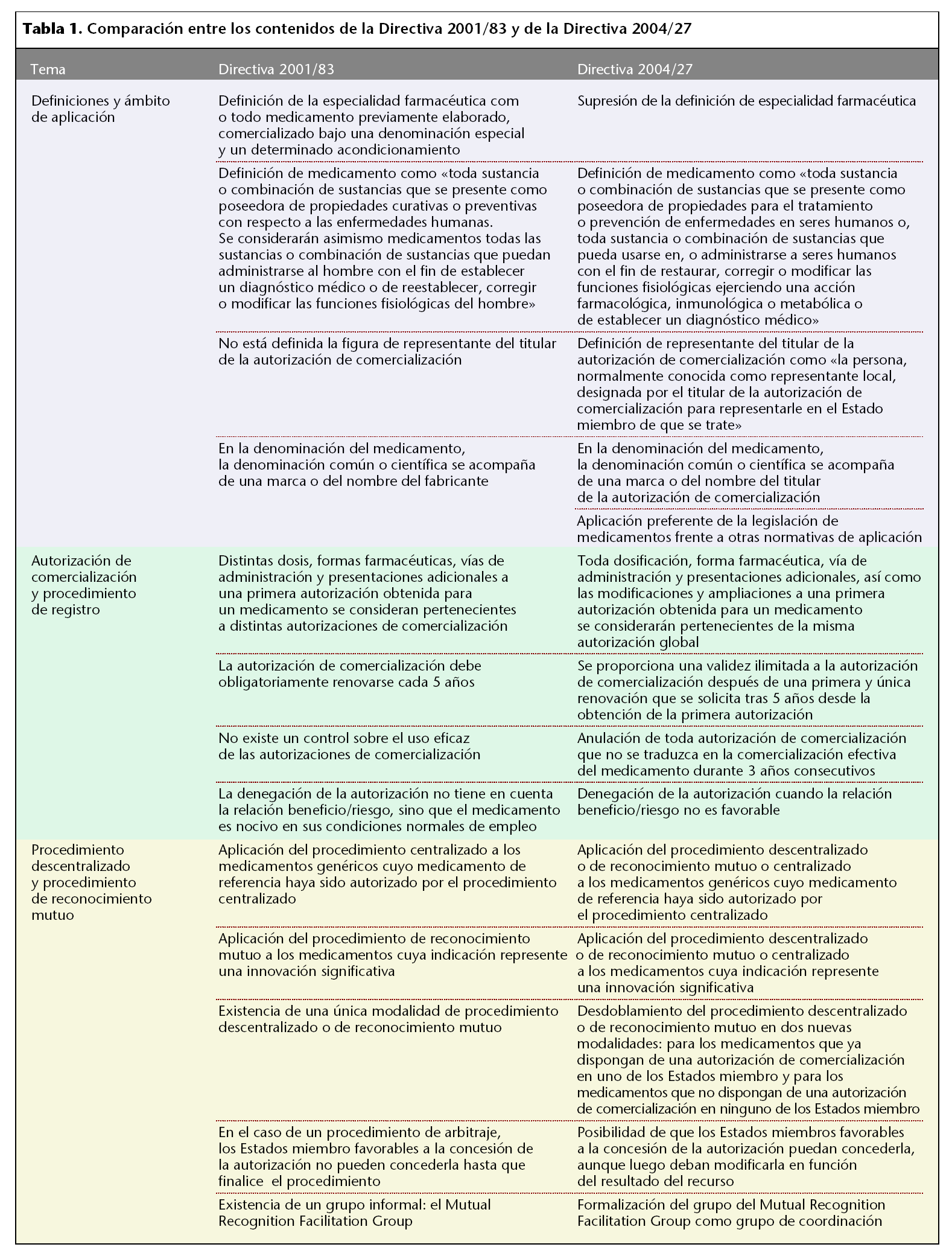
La nueva directiva suprime el término de «especialidad farmacéutica» por no considerarla dentro de su ámbito de aplicación y modifica la definición de «medicamento» para englobar medicamentos de nuevas terapias tales como la terapia génica, los radiofármacos y ciertos medicamentos de uso tópico. Además, la nueva directiva incorpora la figura de representante del titular de la autorización de comercialización, entendiéndose como la persona, normalmente conocida como representante local, designada por el titular de la autorización de comercialización para representarle en el Estado miembro de que se trate. Al titular, con independencia de la designación de un representante, la directiva precisa que le corresponde la responsabilidad de la comercialización del medicamento. En conexión con este punto, también se modifica la definición de denominación del medicamento de manera que en ella pueda figurar el nombre del titular de la autorización de comercialización en lugar del fabricante.
Por último, cabe destacar que para productos frontera será de aplicación preferente la legislación de medicamentos frente a otras normativas de aplicación, por ser ésta más protectora de la salud pública.
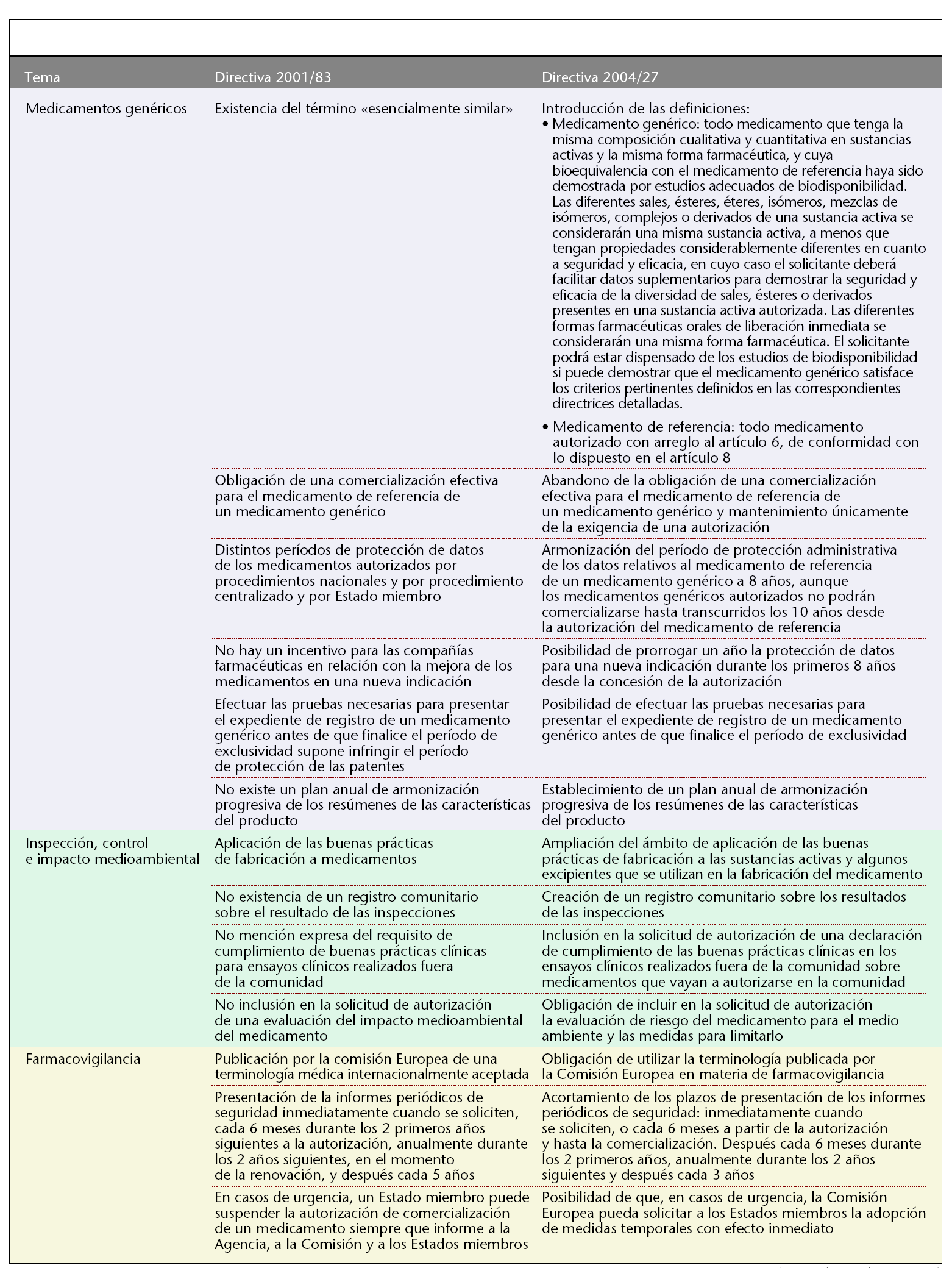
En la tabla 1 se resumen los principales cambios que se han producido.

Autorización de comercialización y procedimiento de registro
La directiva modifica sustancialmente el concepto actual de autorización de comercialización, dándole un carácter unitario de manera que después de la obtención de una primera autorización para un medicamento, toda dosificación, forma farmacéutica, vía de administración y presentación adicionales, o cualquier ampliación o modificación que se produzca y para la que se obtenga también autorización se considerará perteneciente a la misma autorización inicial. Además, una autorización de comercialización pasa a tener una validez ilimitada después de una primera y única renovación que se solicita tras 5 años desde la obtención de la primera autorización. En esta única renovación, la compañía debe presentar un expediente de registro consolidado con todas las variaciones introducidas hasta el momento. En oposición a la validez ilimitada de la autorización, ésta caduca si el medicamento no se comercializa dentro de los 3 primeros años posteriores a la obtención de la autorización, o si se interrumpe la comercialización durante el mismo período.
Por último, se toma la relación beneficio/riesgo como base para la denegación, suspensión o retirada de una autorización de comercialización en lugar de sólo los efectos nocivos.
Procedimiento de registro de reconocimiento mutuo
El cuanto al ámbito de aplicación, el procedimiento de reconocimiento mutuo se amplía a los medicamentos genéricos de medicamentos de referencia autorizados por procedimiento de registro centralizado, que podrán optar por ambos procedimientos. De igual forma, los medicamentos cuya indicación represente una innovación significativa también podrán optar por ambos procedimientos. Además, la directiva revisa el funcionamiento del procedimiento y refuerza la cooperación entre los Estados miembro mediante la incorporación de un procedimiento descentralizado, de 210 días, para los medicamentos que no dispongan de una autorización de comercialización en ninguno de los Estados miembro. Esta cooperación entre los Estados se daría antes de que se adoptara una decisión sobre la base de la evaluación efectuada por uno de ellos. El actual procedimiento de reconocimiento mutuo se restringiría a los medicamentos que ya dispusieran de una autorización de comercialización en uno de los Estados miembros, pero cuyo titular quisiera ampliar su distribución a otros Estados miembro; en ese caso, el plazo se reduciría a 180 días.
Otra novedad se da en los procedimientos de recursos en los que existen discrepancias entre Estados miembro en la concesión de la autorización de comercialización. Además de disminuir su plazo, la directiva dispone que se siga el proceso de recurso aunque los países favorables a conceder la autorización puedan concederla de forma temporal y luego modificarla en función del resultado del procedimiento.
Por último, se crea un Grupo de Coordinación formado por un representante de cada Estado miembro para la gestión de problemas relacionados con la autorización de comercialización en dos a más Estados miembro.
Medicamentos genéricos
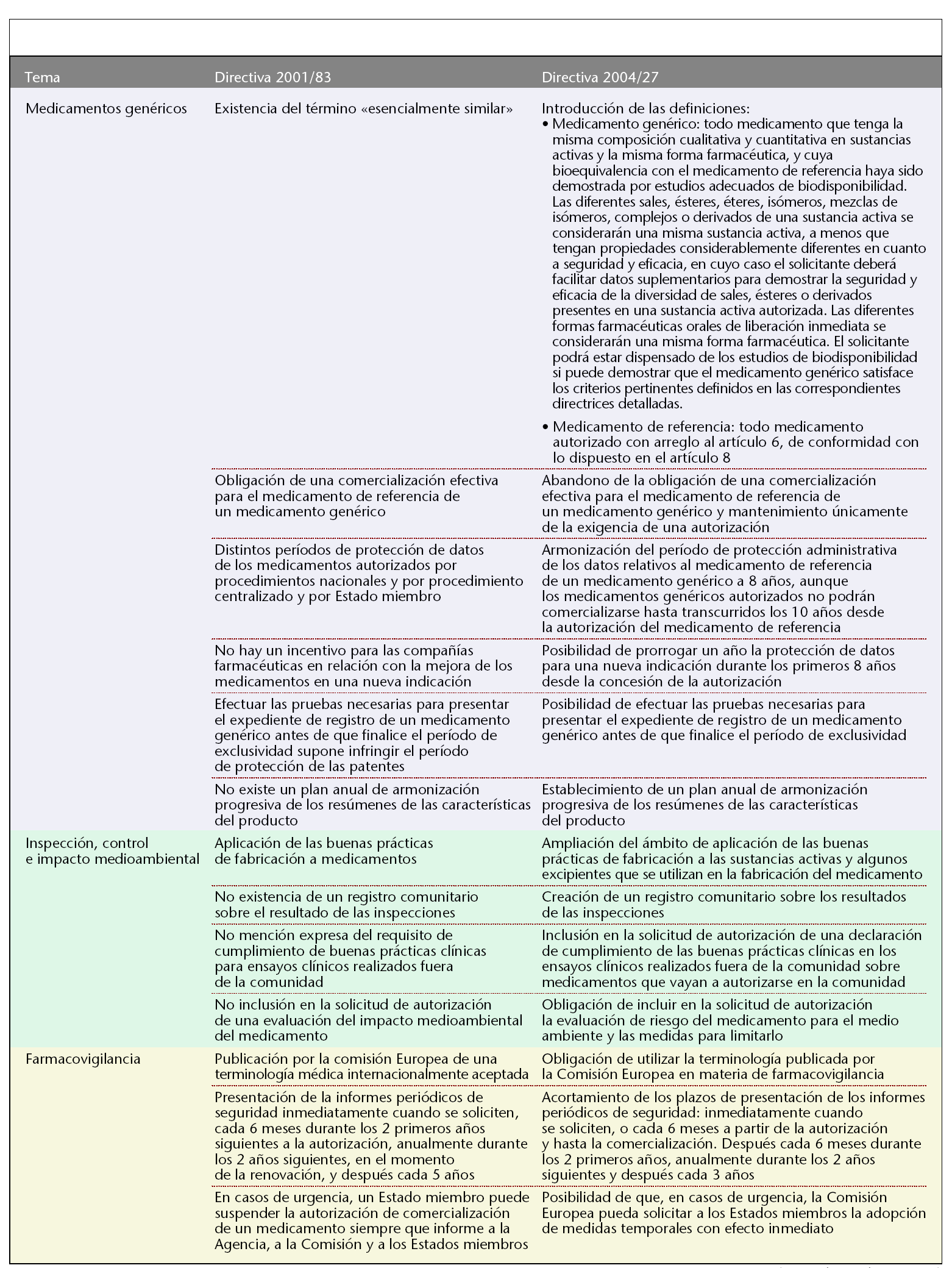
La nueva directiva introduce por primera vez la definición de medicamento genérico y medicamento de referencia (tabla 1). Además, adopta toda una serie de disposiciones con el fin de mejorar las posibilidades de llegada al mercado de los medicamentos genéricos, que en la actualidad constituyen una parte importante del mercado de medicamentos. En primer lugar, se abandona la obligación de que el medicamento de referencia al que se refiere el medicamento genérico se encuentre comercializado en el Estado miembro en el que se cursa la solicitud de autorización. También se eliminan las divergencias que hasta ahora existían en el período de protección administrativa de datos, que dependía del tipo de procedimiento de registro y del Estado miembro. Así, se fija un único período de protección en 8 años, aunque los medicamentos genéricos autorizados no podrán comercializarse hasta transcurridos 10 años desde la autorización del medicamento de referencia. Por el contrario, para favorecer la búsqueda de nuevas indicaciones terapéuticas que presenten un beneficio clínico importante y supongan una mejora del bienestar y la calidad de vida del paciente, se dispone que la compañía farmacéutica se beneficie de hasta un máximo de 1 año suplementario de protección de datos en el caso de nuevas indicaciones terapéuticas autorizadas durante los 8 primeros años del mencionado período de 10 años. A su vez, la directiva introduce la posibilidad de que una compañía farmacéutica realice el desarrollo, las pruebas y la experimentación necesarios para conseguir el registro de un medicamento genérico antes de que finalice el período de protección de la patente del medicamento innovador sin por ello violar la reglamentación relativa a la protección de la propiedad industrial y comercial.
También, se intenta superar el obstáculo de la desarmonización existente entre las fichas técnicas de los medicamentos de referencia en los distintos Estados miembro mediante la creación de un plan de armonización progresivo de fichas técnicas de los medicamentos de referencia.
Por último, destacar la introducción de una disposición en materia de medicamentos biológicos similares a un medicamento de referencia. Para estos medicamentos, que generalmente no suelen reunir todas las condiciones para ser considerados medicamentos genéricos, se dispone que deberán facilitar los resultados de las pruebas adecuadas para satisfacer las condiciones de seguridad y eficacia.
Inspección, control e impacto medioambiental
Para garantizar la calidad de los medicamentos, la directiva amplía el ámbito de aplicación de las inspecciones a principios activos y determinados excipientes exigiéndoles también el cumplimiento de las prácticas de correcta fabricación de los medicamentos. Además, se incrementa la coordinación entre los distintos Estados miembros mediante la creación de un registro comunitario sobre los resultados de las inspecciones.
También se incide en el cumplimiento de las buenas prácticas clínicas en los ensayos clínicos realizados en terceros países sobre medicamentos que vayan a autorizarse en la comunidad.
Por último, la directiva promueve la limitación del impacto medioambiental de los medicamentos, aunque no hasta el punto de ser criterio suficiente para denegar una autorización de comercialización.
Farmacovigilancia
La directiva adopta diferentes disposiciones encaminadas a reforzar la farmacovigilancia. Así, obliga a la utilización de la terminología médica MedDRA aceptada internacionalmente con el fin de facilitar el intercambio y la comprensión de la información de reacciones adversas a medicamentos. Además, se incrementa la frecuencia de presentación de informes periódicos de seguridad de los medicamentos a las autoridades sanitarias. Por último, la directiva también obliga a los Estados miembros a adoptar medidas de urgencia a petición del Comité, con carácter inmediato, previas a las medidas definitivas.
El actual procedimiento de reconocimiento mutuo se restringiría a los medicamentos que ya dispusieran de una autorización de comercialización en uno de los Estados miembro, pero cuyo titular quisiera ampliar su distribución a otros Estados miembro
Información a los pacientes
La directiva prevé para el paciente el establecimiento de un orden lógico de los apartados que obligatoriamente deben figurar en el prospecto. También dispone la obligación de que el nombre del medicamento se indique en alfabeto braille en el envase y que los prospectos, a petición de las organizaciones de pacientes, también se encuentren adaptados a personas invidentes.
Por último, la actual directiva prevé en un plazo de 3 años la elaboración de un informe sobre las prácticas en materia de información (sobre todo en Internet) y los riesgos y beneficios para el paciente, con el fin de definir una estrategia futura de información al paciente.
Plazo de transposición
Los diferentes Estados miembro que integran la Unión Europea están obligados a trasponer todos los dictámenes de la presente directiva a sus ordenamientos jurídicos nacionales antes del 30 de octubre del 2005.