



Estudiados la evolución y el funcionamiento actual del Sistema Español de Farmacovigilancia (SEFV)1 y el papel de la industria farmacéutica como parte de este sistema2, es momento de analizar profundamente la intervención de las autoridades sanitarias cuando se presenta un problema de seguridad de un medicamento.

La actividad del Sistema Español de Farmacovigilancia proporciona información para evaluar la seguridad de los medicamentos que se comercializan en España. En los casos en que se identifica un riesgo real para la salud de las personas, las autoridades sanitarias competentes deben considerar la necesidad de emprender las medidas reguladoras más oportunas y salvaguardar la salud pública.
El RD 711/2002, de 19 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano en España (REF), dedica el capítulo IV a la «Intervención administrativa» (artículos 11 a 17).
Organismos competentes
La Agencia Española del Medicamento (AEM) es el organismo que concede la autorización a un medicamento, previa a su comercialización, basándose en criterios de calidad, eficacia y seguridad. Además, desarrolla funciones específicas en materia de farmacovigilancia concretadas en el artículo 5 del RD 711/2002. De este modo, el real decreto asigna a la AEM la misión de tomar las acciones reguladoras necesarias para asegurar que los medicamentos que se encuentran comercializados continúan siendo de calidad, seguros y eficaces.
En concreto, en materia de seguridad, la AEM desarrolla sus funciones a través de dos estructuras básicas:
La División de Farmacoepidemiología y Farmacovigilancia de la Subdirección General de Seguridad de Medicamentos.
El Comité de Seguridad de Medicamentos de Uso Humano.
El Comité de Seguridad de Medicamentos de Uso Humano es un órgano de asesoramiento técnico y científico de la División de Farmacoepidemiología y Farmacovigilancia. En materia de intervención administrativa ante un problema de seguridad, el Comité informa preceptivamente a la División en el procedimiento de modificación, suspensión o revocación de una autorización de comercialización de medicamentos de uso humano. Aunque estos informes nunca son vinculantes, constituyen la base de la toma de decisiones de la División.
Tipos de acciones reguladoras
Las acciones reguladoras en las que puede derivar una decisión de la AEM ante un problema de seguridad de un determinado medicamento son las siguientes:
Modificación de las condiciones de uso terapéutico que se autorizaron por parte de la AEM para el medicamento en estudio.
Suspensión o revocación de la autorización del medicamento en estudio.
Modificación de las condiciones de uso terapéutico
De acuerdo con el artículo 12 del RD 711/2002, la AEM puede restringir las condiciones de autorización o dictaminar una restricción en su ámbito de uso.
En el primer caso, la modificación de las condiciones de autorización se traduce, por ejemplo, en la inclusión de advertencias, precauciones o nuevas contraindicaciones en el prospecto o en la ficha técnica. Los textos y demás características de la ficha técnica, prospecto y etiquetado forman parte de la autorización de comercialización de la especialidad farmacéutica y, por tanto, cualquier modificación requiere también autorización previa por parte de las autoridades sanitarias competentes.
Los textos y demás características de la ficha técnica, prospecto y etiquetado forman parte de la autorización de comercialización de la especialidad farmacéutica
En el segundo caso, la restricción del uso del medicamento puede traducirse en la obligación de que convierta en un medicamento de uso hospitalario, en un medicamento de diagnóstico hospitalario o de prescripción por médicos especialistas, o un medicamento de especial control.
Suspensión o revocación de la autorización
El artículo 13 del RD 711/2002, en desarrollo de la Ley 25/1990 de la Ley del Medicamento (LM), establece que en una de las siguientes condiciones la AEM puede dictaminar la suspensión temporal o bien la retirada definitiva del medicamento:
El medicamento resulta nocivo o no seguro en las condiciones normales de empleo (artículo 26.a) de la LM).
El medicamento resulta no ser terapéuticamente eficaz (artículo 26.b) de la LM).
El medicamento, por cualquier otra causa, supone un riesgo previsible para la salud o seguridad de las personas (artículo 26.i) de la LM).
El medicamento muestra una relación beneficio/riesgo desfavorable (artículo 10.3) de la LM).
Hasta que se resuelva el procedimiento de suspensión o revocación por motivos de salud pública puede suspenderse preventivamente el medicamento, supuesto en que el medicamento debe retirarse del mercado bajo la responsabilidad de la compañía farmacéutica titular.
Procedimiento de intervención administrativa
El procedimiento de intervención administrativa puede explicarse en seis etapas (fig. 1).
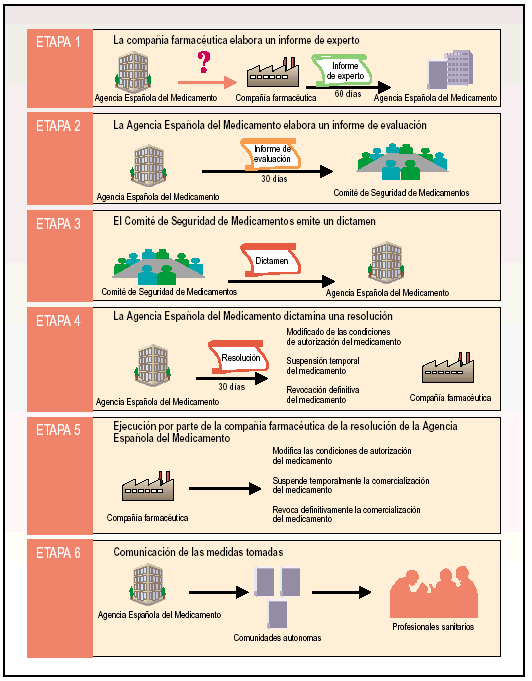
Fig. 1. Etapas en el procedimiento de intervención administrativa ante un problema de seguridad con un medicamento.
Etapa 1. La compañía farmacéutica elabora un informe de experto
La AEM solicita a la compañía farmacéutica titular de la autorización del medicamento un informe redactado por un experto. Este informe, que se debe elaborar en un plazo máximo de 60 días desde la solicitud por parte de la AEM, estará compuesto por una evaluación de la relación beneficio/riesgo del medicamento y por una propuesta de medidas a tomar para reducir el riesgo de inseguridad del medicamento.
Etapa 2. La AEM elabora un informe de evaluación
Basándose en el informe de experto recibido de la compañía farmacéutica titular, la AEM elabora, en un plazo máximo de 30 días, un informe de evaluación del problema de seguridad que presenta el medicamento. Una vez elaborado este informe, la AEM lo remite al Comité de Seguridad de Medicamentos.
Etapa 3. El Comité de Seguridadde Medicamentos emite un dictamen
Fundado en el informe de evaluación recibido de la AEM, el Comité de Seguridad de Medicamento emite un dictamen preceptivo para la Agencia. El dictamen del Comité de Seguridad de Medicamentos no es en ningún caso vinculante.
Etapa 4. La AEM dictaminauna resolución
Basándose en el dictamen emitido por el Comité de Seguridad de Medicamentos, la AEM, dispone de máximo 30 días para dictaminar una resolución al problema de inseguridad del medicamento y comunicarla a la compañía farmacéutica titular.
En los casos en que el medicamento esté autorizado por un procedimiento de reconocimiento mutuo en distintos países de la Unión Europea, antes de que la AEM dictamine una resolución, se debe someter el problema de inseguridad al Comité de Especialidades Farmacéuticas de la Agencia Europea para la Evaluación de Medicamentos para que se pueda tomar una decisión global a escala comunitaria. Una vez el Comité de Especialidades Farmacéuticas notifique la decisión comunitaria a la AEM, ésta dispone de máximo 30 días para notificar la resolución a la compañía farmacéutica.
La resolución emitida por la AEM puede provocar la modificación de las condiciones de autorización del medicamento, la suspensión temporal del medicamento o la revocación definitiva del medicamento.
La compañía farmacéutica puede presentar un recurso contra cualquiera de las decisiones tomadas por la AEM.
Cuando la resolución de la AEM disponga una modificación en las condiciones de autorización del medicamento, esta resolución indicará específicamente los cambios que la compañía farmacéutica debe realizar en la ficha técnica, en el prospecto, en el etiquetado y/o en el ámbito de uso del medicamento, además de otras medidas de información del riesgo e información a profesionales sanitarios y pacientes.
Cuando la resolución de la AEM disponga una suspensión, revocación o modificación relevante en el uso del medicamento, debe informar a las comunidades autónomas, a la Agencia Europea para la Evaluación de Medicamentos, a los demás estados miembros y a la compañía farmacéutica titular de la autorización de comercialización del medicamento. Además, si puede afectar a la salud de terceros países, se debe también poner en conocimiento de la Organización Mundial de la Salud.
Etapa 5. Ejecución por parte de la compañía farmacéutica de la resolución de la AEM
Cuando la compañía farmacéutica deba proceder a la modificación de las condiciones de autorización del medicamento, seguirá las disposiciones del RD 767/93, de 21 de mayo y la Circular 18/97 de la Dirección General de Farmacia y Productos Sanitarios sobre modificaciones de las condiciones de autorización de las especialidades farmacéuticas de uso humano. Como excepción, los medicamentos autorizados por el procedimiento de registro centralizado deben proceder de acuerdo al Reglamento 2.309/93 del Consejo, de 22 de julio.
En general, para proceder a modificar las condiciones de autorización del medicamento, la compañía farmacéutica titular presenta, junto a la solicitud, la documentación técnica necesaria de acuerdo a lo especificado en la Circular 18/97 y el pago de las correspondientes tasas. Las autoridades sanitarias estudian y evalúan la documentación presentada, sobre la que conceden o deniegan la variación solicitada.
La División de Farmacoepidemiología y Farmacovigilancia de la AEM es responsable de la evaluación de las modificaciones relacionadas con la farmacovigilancia de los medicamentos. Por ello, la compañía farmacéutica debe presentar la documentación por duplicado al registro central de la AEM dirigida a la División de Farmacoepidemiología y Farmacovigilancia.
De acuerdo con la Circular 15/2002, de 30 de septiembre, relativa a los procedimientos de comunicación en materia de farmacovigilancia de medicamentos de uso humano, entre la Industria Farmacéutica y el Sistema Español de Farmacovigilancia de medicamentos de uso humano, las variaciones relacionadas con la farmacovigilancia son las siguientes:
Variaciones de importancia menor
Variación tipo I n.º 9. Supresión de una indicación.
Variaciones de importancia mayor
Variación tipo II n.º 112. Eliminación de reacciones adversas.
Variación tipo II n.º 113. Modificación no restrictiva sobre la sobredosificación.
Variación tipo II n.º 230. Adición de contraindicaciones, advertencias, precauciones interacciones, reacciones adversas en la ficha técnica.
Variación Tipo II n.º 231. Modificación restrictiva sobre el embarazo y la lactancia, estado de alerta y la sobredosificación en la ficha técnica.
La persona de contacto de la compañía farmacéutica con la División de Farmacoepidemiología y Farmacovigilancia para aclaraciones técnicas sobre las variaciones debe ser siempre el responsable de farmacovigilancia de la compañía.
Etapa 6. Comunicación de las medidas tomadas para el medicamento
La AEM debe informar de las medidas tomadas para el medicamento de que deban ser conocedores los profesionales sanitarios. La comunicación se ejerce de la AEM a las distintas comunidades autónomas y otros organismos implicados en salud pública, y de las distintas comunidades autónomas a los profesionales sanitarios, tanto del sector público como privado.
A su vez, tanto la AEM como las comunidades autónomas tienen la responsabilidad de informar a los ciudadanos, de la forma más apropiada, de los riesgos asociados a los medicamentos.
El Sistema de Farmacovigilancia para la identificación de los problemas de seguridad de los medicamentos no tendría sentido sin un sistema de intervención sanitaria eficaz
Conclusión
Podemos concluir subrayando que el Sistema de Farmacovigilancia para la identificación de los problemas de seguridad de los medicamentos no tendría sentido sin un sistema de intervención sanitaria eficaz en cuanto se detecta un problema real de inseguridad en el mercado. El alcance de esta actuación repercute directamente en la industria farmacéutica titular del medicamento y sus consecuencias están en directa relación con la gravedad del problema.