


Alglucosidasa alfa es el primer y único tratamiento específico para la enfermedad de Pompe, también conocida como déficit de maltasa ácida o glucogenosis tipo II, una dolencia neuromuscular debilitante, y a veces mortal, que se caracteriza por una degeneración progresiva esquelética, respiratoria y de los músculos cardíacos. Los pacientes que presentan esta enfermedad rara tienen un déficit de la enzima alfaglucosidasa ácida (GAA) que conduce a una acumulación progresiva de glucógeno en el lisosoma. Alglucosidasa alfa trata la causa subyacente de la enfermedad y reemplaza la enzima deficitaria.
La enfermedad de Pompe es una enfermedad rara que se caracteriza por causar una dolencia neuromuscular progresiva, debilitante y, a veces, mortal, que comprende toda una serie de fenotipos que abarcan desde una rápida progresión que suele ser fatal antes del primer año de vida hasta una progresión más lenta que deriva en una significativa morbilidad y/o mortalidad prematuras.
La causa de esta enfermedad es el déficit de la enzima GAA que deriva en una acumulación de glucógeno en los lisosomas, con graves consecuencias en las células cardíacas, esqueléticas, respiratorias y del músculo liso. La gravedad de la enfermedad depende del grado de déficit enzimático.
Antes de la terapia de sustitución enzimática, el tratamiento de la enfermedad de Pompe era paliativo y sintomático y no abordaba la causa subyacente de la enfermedad.
La función de la GAA es catalizar la degradación del glucógeno a glucosa dentro de los lisosomas; por tanto, la base patológica de la enfermedad de Pompe es el depósito lisosomal de glucógeno debido a una disminución o falta de actividad de la GAA.

Mecanismo de acción
Alglucosidasa alfa reemplaza la enzima deficitaria en la enfermedad de Pompe y es el primer y único tratamiento que aborda la causa subyacente de la enfermedad. La secuencia de aminoácidos del fármaco es idéntica a la forma natural de GAA humana. Al igual que la enzima original, alglucosidasa alfa degrada el glucógeno y cataliza la hidrólisis de los enlaces glucosídicos alfa-1.4 y alfa-1.6 de glucógeno lisosomal. De este modo, la terapia de sustitución enzimática compensa el déficit de GAA que causa la enfermedad de Pompe. La administración del fármaco se realiza mediante infusiones bisemanales.
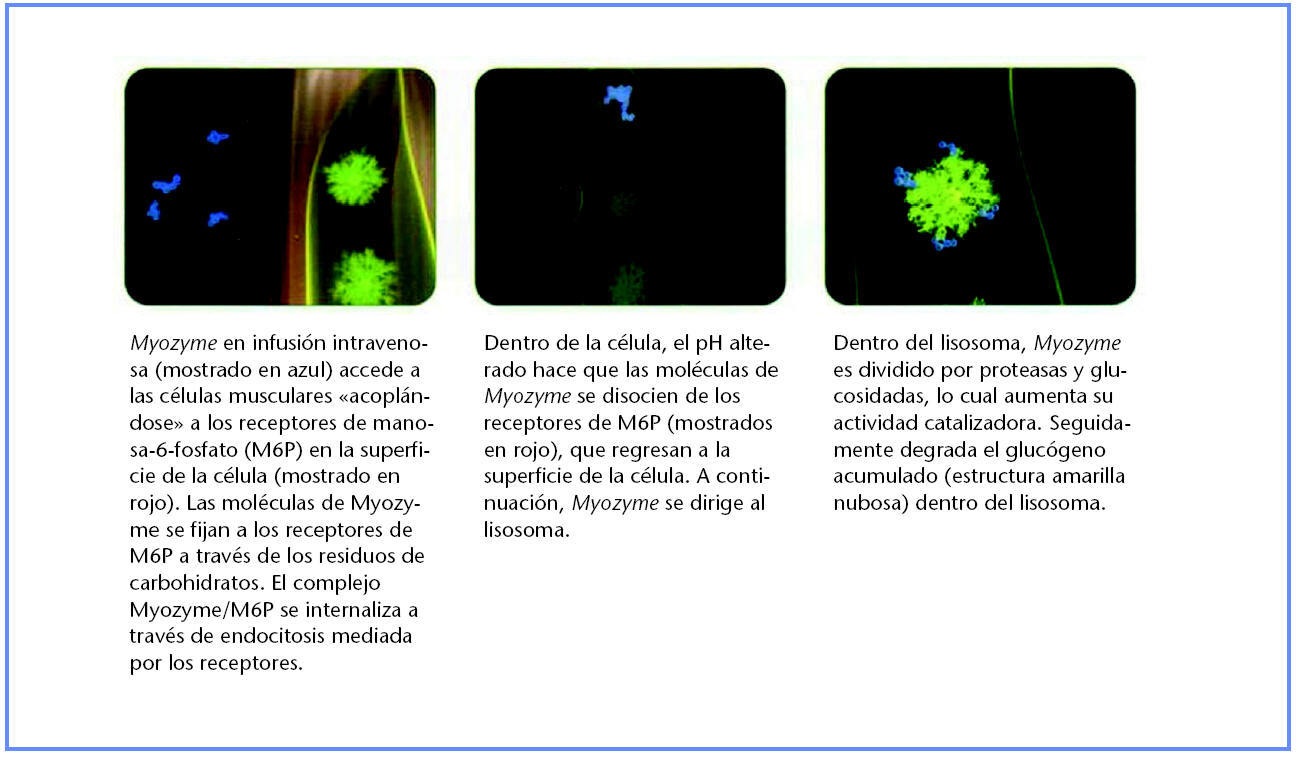
Tras la administración de alglucosidasa alfa, al igual que su forma natural humana GAA, el fármaco se dirige al lisosoma mediante los residuos de manosa-6-fosfato (M6P) en la superficie de la molécula proteínica (30). La infusión de alglucosidasa alfa se capta desde la circulación y se lleva a las células mediante los receptores M6P en la superficie celular. Una vez dentro de la célula, se dirige al lisosoma mediante el mismo proceso de endocitosis mediada por receptores y, una vez en su interior, experimenta una división proteolítica que aumenta su actividad enzimática.
Biotecnología
La producción de alglucosidasa se efectúa siguiendo los métodos más modernos en biotecnología. Este fármaco se produce en células de ovario de hámster chino (CHO). Estas células han sido una base clásica para la fabricación de biofármacos en todo el mundo desde hace más de 10 años.
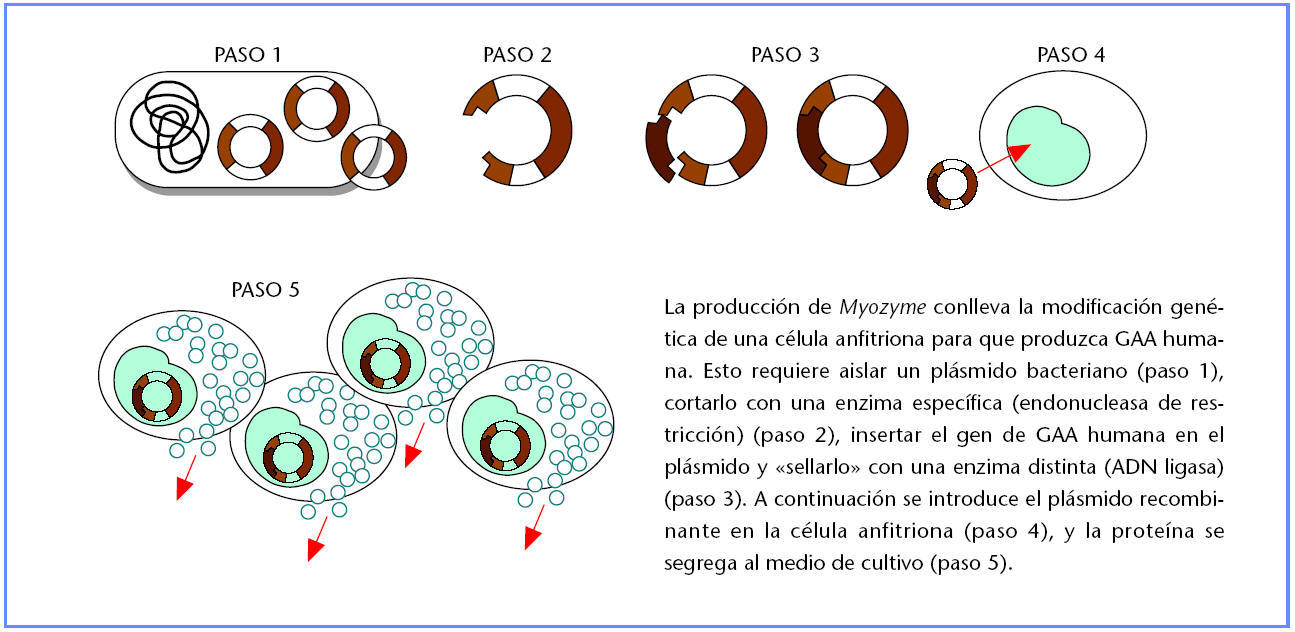
El proceso de fabricación de alglucosidasa alfa empieza con la modificación genética de una célula anfitriona para producir GAA humana. Primero se aísla el gen de GAA humana y se une a un plásmido bacteriano (pequeño anillo aurorreplicante de ADN). Para ello se debe cortar el plásmido con un tipo de enzima (endonucleasa de restricción), insertar el gen de GAA y sellarlo dentro del plasma con otra enzima, la ADN ligasa. A continuación, se inserta el plásmido que contiene GAA en una célula de CHO que actúa como anfitriona para fabricar la proteína.
En un biorreactor en condiciones controladas, las células CHO genéticamente modificadas se desarrollan en un medio líquido enriquecido con nutrientes y segregan GAA al medio. La GAA humana recombinante se aísla del medio de cultivo utilizando un riguroso proceso de purificación de múltiples pasos que incluye cromatografía y nanofiltración capaz de eliminar partículas víricas.
Para cumplir con las reglamentaciones internacionales, el producto se testa rigurosamente en cada etapa de su elaboración. El producto final se formula con manitol para mejorar la estabilidad, se filtra en condiciones estériles, se introduce en viales y se liofiliza para garantizar la estabilidad. Tras la liofilización, el producto se somete a rigurosos controles de calidad.
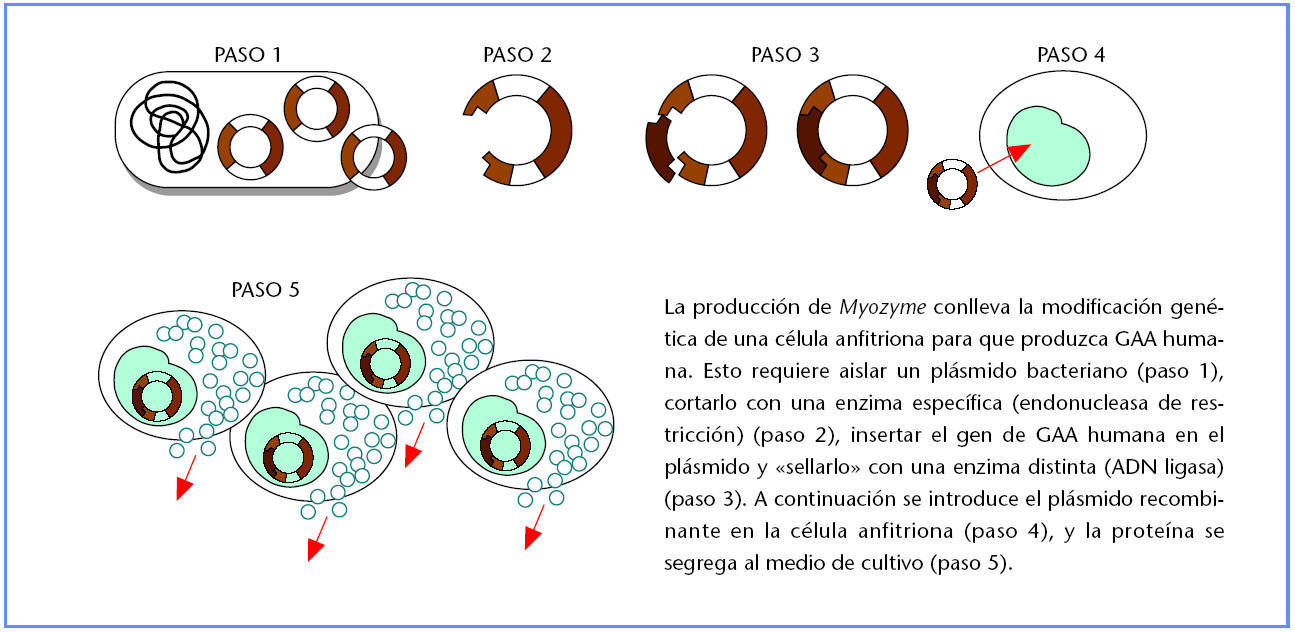
Producción de proteínas utilizando tecnología de ADN recombinante
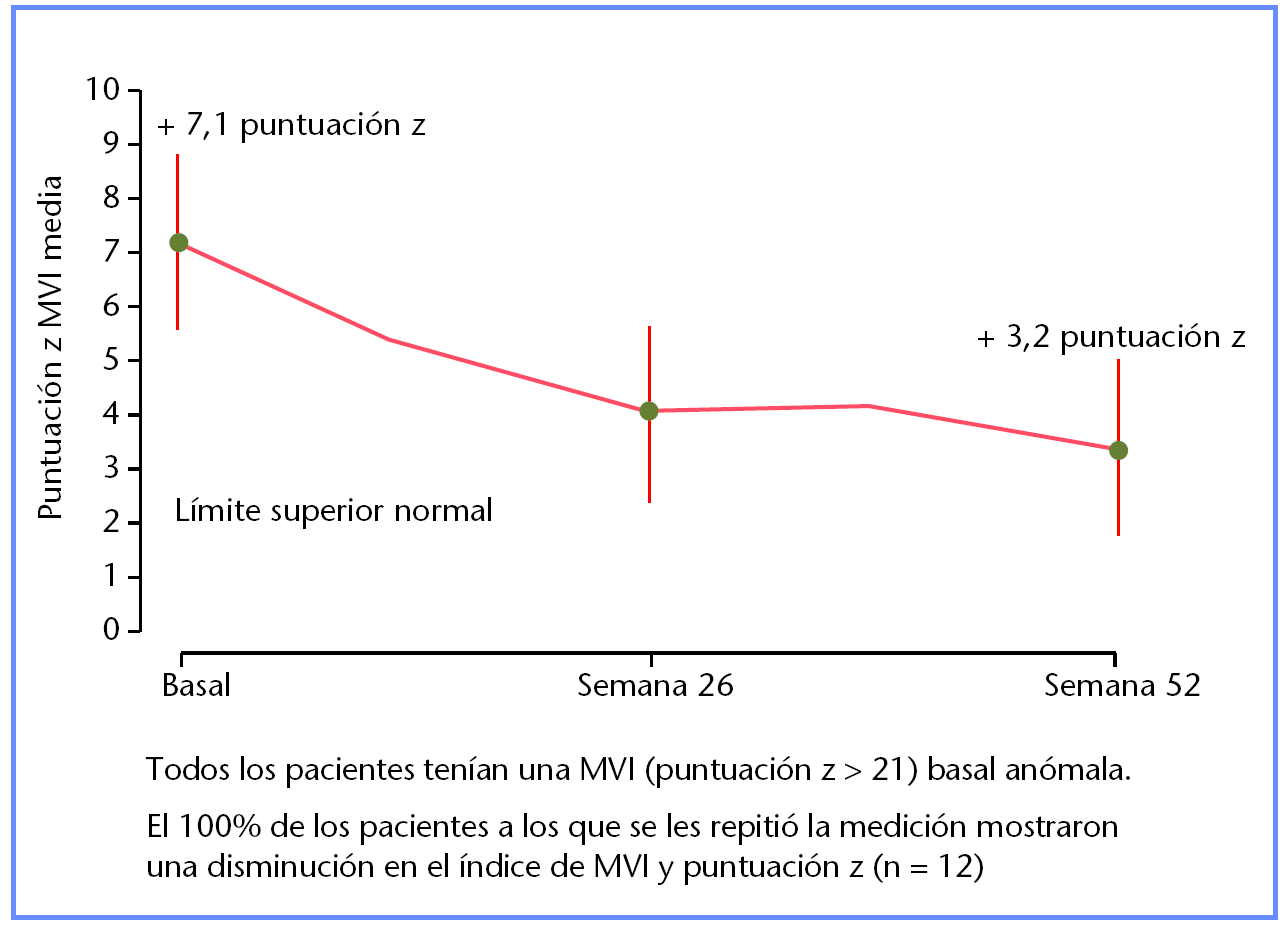
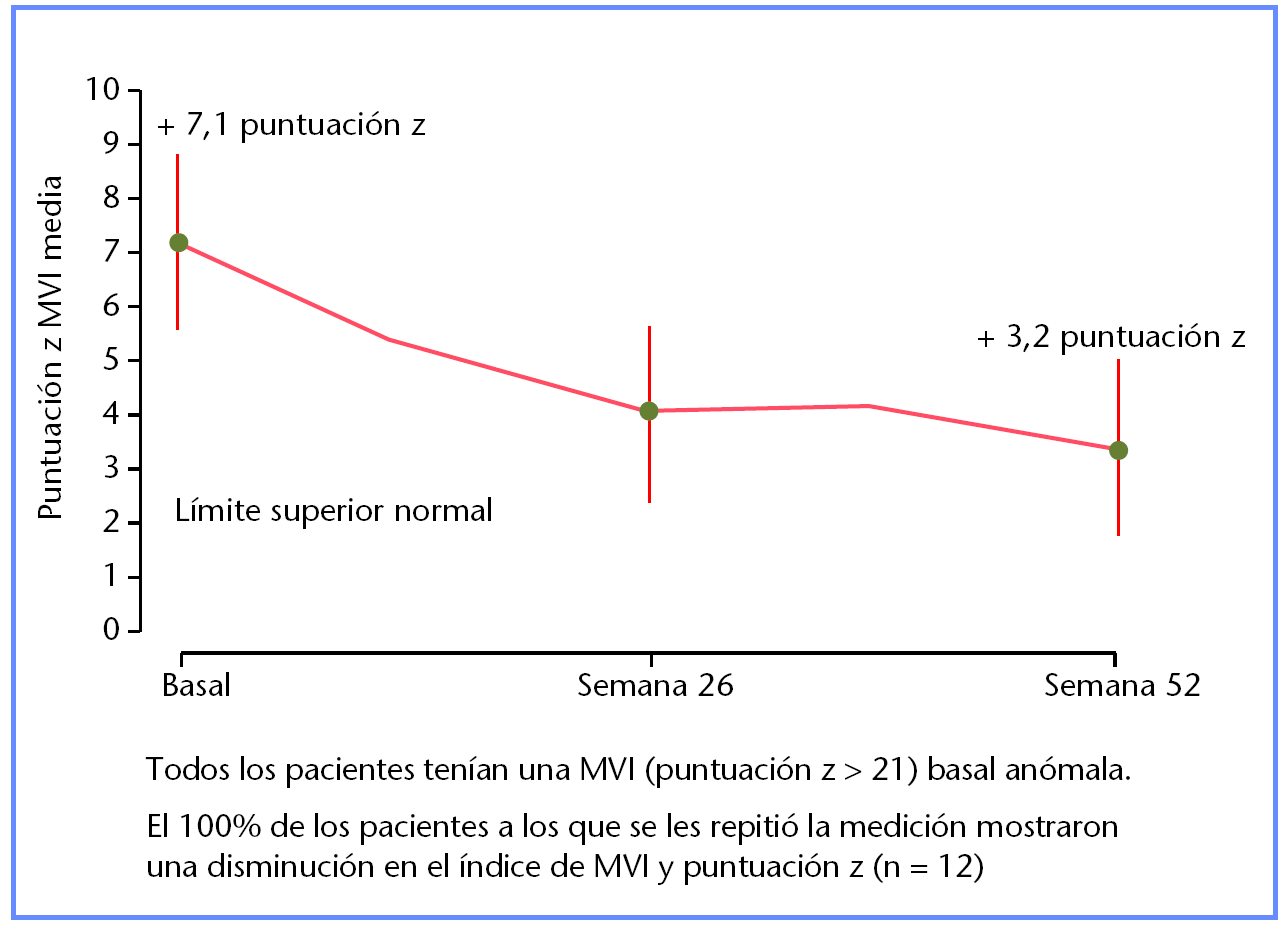
Mejora de miocardiopatía medida por la disminución en la masa del ventrículo izquierdo en 12 bebés menores de 6 meses al inicio del tratamiento con alglucosidasa alfa
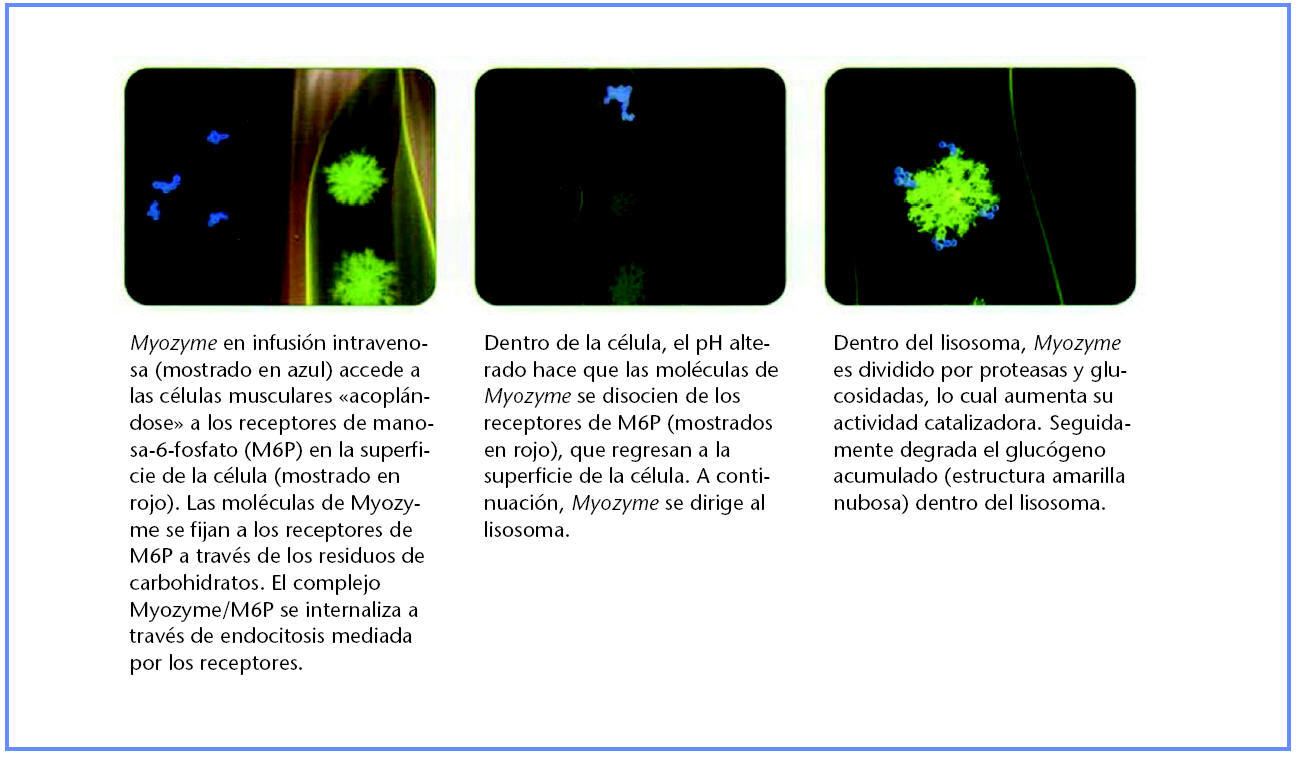
Mecanismo de acción de alglucosidasa alfa
Eficacia clínica
Los ensayos clínicos en bebés y niños pequeños han demostrado que alglucosidasa alfa aumenta la supervivencia en comparación con pacientes no tratados. Además, el fármaco consigue mejorar la función cardíaca y estabilizar o mejorar los parámetros de crecimiento. Sin embargo, la respuesta motora y respiratoria al tratamiento ha sido más variable. Los datos proponen la necesidad de un diagnóstico temprano para conseguir mejores resultados.
En niños mayores y adultos, los datos de los ensayos sugieren que alglucosidasa alfa puede reducir la dependencia de la ventilación mecánica, mejorar la movilidad, aumentar la calidad de vida y prevenir o retrasar la progresión de la enfermedad. La enfermedad de Pompe es progresiva; por lo tanto, una mejoría o bien la estabilización de la enfermedad pueden considerarse efectos terapéuticos.
Seguridad
En algunas ocasiones se han observado reacciones anafilácticas que pueden ser mortales, especialmente en pacientes en los que la enfermedad de Pompe se presentó tardíamente. En el momento de la administración de alglucosidasa alfa, se deberá disponer de tratamientos médicos adecuados, debido al potencial de reacciones graves. En ese caso, deberá interrumpirse el tratamiento.
En los estudios clínicos se describen las siguientes reacciones adversas: erupciones, enrojecimiento, urticaria, pirexia, tos, taquicardia, disminución de la saturación de oxígeno, vómitos, taquiapnea, agitación, elevación de la tensión arterial, cianosis, hipertensión, irritabilidad, palidez, prurito, arcadas, rigidez, temblores, hipotensión, broncoespasmos, eritema, edema facial, sensación de calor, cefalea, hiperhidrosis, aumento de la lagrimación, livedo reticular, náuseas, edema periorbital, inquietud y sibilancia.
En bebés, las reacciones adversas más frecuentes fueron urticaria, enrojecimiento, pirexia y erupción.
La mayoría de los pacientes desarrollaron anticuerpos IgG contra alglucosidasa alfa; a los 3 meses de tratamiento, el nivel de reacción de los anticuerpos debe monitorizarse regularmente.
Conclusión
Alglucosidasa alfa es el primer y único tratamiento específico para la enfermedad de Pompe, una enfermedad de alto impacto familiar y social, progresiva y que puede tener consecuencias fatales.
Es preciso un diagnóstico temprano para poder iniciar el tratamiento antes de que ocurran daños musculares irreversibles.
Los ensayos clínicos en bebés y niños pequeños muestran que el fármaco prolonga significativamente la supervivencia y puede detener o retrasar la progresión de la enfermedad al sustituir la enzima deficiente.
En niños y adultos, el fármaco ha demostrado su eficacia en reducir o estabilizar el uso de un respirador, así como un aumento de las mejoras funcionales.