


EL SR. W., UN VARÓN BLANCO de 55 años de ascendencia europea, se presenta en un centro de atención primaria con síntomas poco precisos de cansancio inexplicable, mialgia, molestias abdominales y dolor de rodilla bilateral. Entre sus antecedentes familiares pueden citarse la muerte de su padre por infarto de miocardio a los 53 años, la de una hermana por carcinoma hepatocelular a los 45 años y la de un hermano a los 52 años, al cual se le detectaron indicios de sobrecarga férrica en la autopsia. Otra hermana del paciente, de 42 años, aparentemente se encuentra en buen estado. El Sr. W. comenta que bebe una copa de vino cada noche durante la cena y que no se le ha recetado ningún medicamento ni toma vitaminas de venta sin receta ni productos de fitoterapia o suplementos nutricionales; tampoco drogas.
Entre los resultados de la evaluación física pueden citarse hepatomegalia leve, aspecto bronceado de la piel e inflamación de las articulaciones metacarpofalángicas. Mide 1,83 m y pesa 90,7 kg. El índice de masa corporal es 27,1, lo que indica que el Sr. W. tiene sobrepeso. Los signos vitales son: PA de 118/80mmHg; temperatura de 37°C; pulso apical normal de 86 latidos/minuto, y 20 respiraciones/minuto.
Los resultados de los análisis de sangre del Sr. W. muestran un nivel de ferritina sérica de 1.048 ng/ml (valores normales en hombres: de 18 a 270 ng/ml) y saturación de transferrina del 95% (valores normales en hombres:del 10% al 50%)1.El diagnóstico preliminar es hemocromatosis hereditaria (HH), un trastorno genético bastante frecuente en adultos2.
La capacidad de prever los cuidados enfermeros que este paciente necesitará permite un tratamiento más oportuno de sus síntomas y signos, así como la prevención de complicaciones. Además de la fisiopatología de la HH, este artículo expone su diagnóstico y tratamiento, junto con importantes temas de enseñanza que las enfermeras deben abordar con los pacientes y sus familias.
¿Qué es la hemocromatosis hereditaria?Conocida como un trastorno por sobrecarga férrica, la HH es una enfermedad genética que aparece como resultado de un aumento de la absorción intestinal de hierro. Los síntomas y signos están relacionados con depósitos de hierro excesivos en los tejidos y órganos, como la piel, el corazón, el hígado, el páncreas y las articulaciones3. Aunque la HH puede tratarse eficazmente en sus primeras etapas, puede afectar gravemente a algunos órganos si el diagnóstico y el tratamiento se retrasan4. Por desgracia, la enfermedad a menudo no se diagnostica durante las primeras etapas puesto que la presentación clínica inicial es imprecisa y muchos médicos no están familiarizados con las enfermedades genéticas5.
La HH se clasifica en varios tipos, basados en factores como la mutación genética implicada y la aparición de los síntomas. Se han identificado más de cuatro tipos, pero estos son los más frecuentes6:
- •
HFE (tipo I o HH clásica): causada por mutación del genHFE; es el tipo más frecuente; herencia autosómica recesiva; los síntomas se inician en la edad adulta.
- •
Hemocromatosis juvenil (tipo II): mutaciones en la hemojuvelina (HJV) en el tipo IIA y en la hepcidina (HAMP) en el tipo IIB; herencia autosómica recesiva; se inicia en la infancia o en la juventud.
- •
Mutación del receptor 2 de transferrina (tipo III): defecto en el receptor 2 de la transferrina; herencia autosómica recesiva; los síntomas se inician en la edad adulta joven.
- •
Mutaciones de ferroportina (tipo IV): mutaciones en un gen que codifica la ferroportina (SLC40A1); dos fenotipos diferentes, de tipo hepático y tipo macrófago; herencia autosómica dominante; se inicia en la edad adulta.
Puesto que la HH se debe más frecuentemente a mutaciones en el gen HFE (tipo I)3, el artículo se centrará en este tipo y a él se referirá como HH.
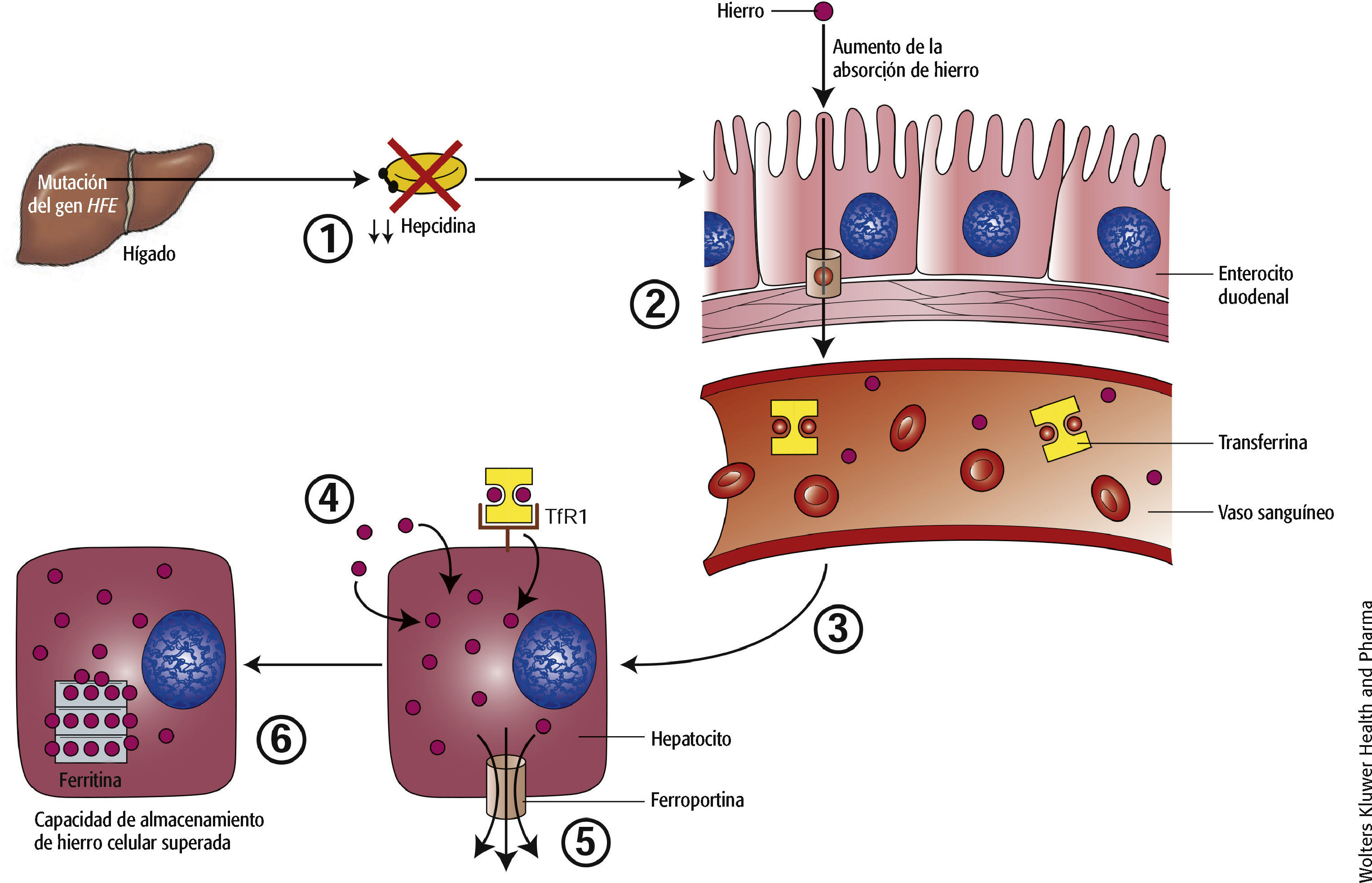
1. La anomalía genética en la hemocromatosis hereditaria (HH) provoca la disminución de la producción hepática de hepcidina. La hepcidina se considera la “principal” hormona reguladora del hierro. El hígado produce hepcidina y esta determina la cantidad de hierro que se absorbe de la dieta y se libera en el cuerpo desde los lugares de almacenamiento. 2. A consecuencia de ello, cuando el hierro de la luz duodenal entra en los enterocitos, no puede regular de manera adecuada su exportación a través de la ferroportina (una proteína especializada, fundamental para la exportación adecuada de hierro de las células) y se absorbe hierro en exceso y se transporta hasta la sangre a través de los enterocitos. 3. Normalmente, el hierro se transporta en la sangre unido a la transferrina y por lo general una tercera parte de esta se satura con hierro. En la HH, no solo se satura la capacidad portadora de hierro de la transferrina (100%), sino que el hierro libre (no unido a la transferrina) también es abundante en la sangre. 4. El hierro entra en los hepatocitos como hierro unido a la transferrina y como hierro libre. El hierro unido a la transferrina entra a través de la vía del receptor 1 de la transferrina (TfR1). El hierro libre entra a través de una vía diferente, que no se conoce con exactitud. 5. A falta de inhibición por parte de la hepcidina, la exportación de hierro por parte de la ferroportina es muy activa. 6. Sin embargo, probablemente a causa de la entrada abrumadora de hierro libre en hepatocitos, la capacidad de almacenamiento y exportación de hierro están superadas, y se acumula un exceso de hierro.
La mutación genética más frecuente que causa la HH, un trastorno autosómico recesivo, es una mutación en el gen HFE (de hereditario y Fe [hierro]) con homocigosis de la mutación C282Y en el cromosoma 66–10. Esta mutación C282Y desactiva el mecanismo de regulación de la absorción intestinal de hierro.
Una persona debe adquirir dos genes mutados (uno de cada progenitor, homocigosis) para desarrollar HH (v. Glosario de términos genéticos). Si una persona hereda solo una copia del gen mutado (heterocigosis), será portadora, por lo general sin síntomas ni signos; una copia del gen no afectado normalmente es suficiente para regular la absorción de hierro. Como “portador asintomático”, esta persona podría transmitir el gen mutado a un hijo. Si ambos progenitores son portadores, cada hijo tiene una probabilidad del 25% de heredar dos copias del gen mutado y probablemente desarrollará el trastorno en la edad adulta9.
Penetrancia se refiere a la proporción de personas en una población que tiene el genotipo y el correspondiente fenotipo o que presenta síntomas de la enfermedad11. En la HH, la penetrancia es incompleta o inferior al 100%, lo que significa que no todos los individuos que tienen la mutación genética presentarán los síntomas. Actualmente se continúa investigando la relación entre la mutación genética y la acumulación de hierro12.
La población étnica más afectada por HH es la blanca y las personas con ascendentes del norte de Europa presentan el riesgo más elevado. En Estados Unidos, la HH afecta a cerca de un millón de personas9.
En términos evolutivos, tener HH quizá alguna vez fue una ventaja genética. Las mujeres heterocigóticas (que tenían un gen mutado y un gen no afectado) pueden haber contado con una ventaja reproductiva porque eran menos propensas a desarrollar anemia ferropénica y estos hombres y mujeres, en términos de supervivencia, habrían tenido una posición ventajosa en tiempos de hambre8.
Alelo: una de dos o más versiones del gen responsable de una variabilidad hereditaria. Las personas heredan dos alelos en cada gen, uno de cada progenitor. Si los alelos son diferentes, el alelo dominante se expresará y el efecto del alelo recesivo estará enmascarado. Para desarrollar una enfermedad genética recesiva, un individuo debe heredar dos copias del alelo mutado.
Autosoma: cualquiera de los cromosomas numerados, en lugar de los cromosomas sexuales. Los seres humanos tienen 22 pares de autosomas y un par de cromosomas sexuales (X e Y). Los autosomas se numeran más o menos en relación con su tamaño, es decir, el cromosoma 1 tiene aproximadamente 2.800 genes, mientras que el cromosoma 22 tiene aproximadamente 750 genes.
Autosómico dominante: patrón de herencia característico de algunas enfermedades genéticas. Autosómico significa que el gen en cuestión se encuentra en uno de los cromosomas numerados o no sexuales. Dominante significa que una sola copia de la mutación asociada a la enfermedad es suficiente para causar la enfermedad. Esto es diferente en un trastorno recesivo, donde se necesitan dos copias de la mutación que causa la enfermedad. La enfermedad de Huntington es un ejemplo de trastorno genético autosómico dominante.
Cromosoma: paquete organizado de ADN que se encuentra en el núcleo de la célula. Los diferentes organismos tienen diferente número de cromosomas. Los seres humanos tienen 23 pares de cromosomas, 22 pares de cromosomas numerados, denominados autosomas, y un par de cromosomas sexuales, X e Y. Cada progenitor contribuye con un cromosoma a cada par de modo que la descendencia obtiene la mitad de sus cromosomas de la madre y la otra mitad, del padre.
Fenotipo: rasgos observables en un individuo, como la altura, el color de los ojos o el grupo sanguíneo. La contribución genética al fenotipo se denomina genotipo. Este determina en gran parte algunas características, mientras que otros rasgos están determinados en gran medida por factores ambientales.
Gen: una secuencia de ADN que ocupa una ubicación específica en un cromosoma; se considera la unidad básica de la herencia.
Genética: estudio de un gen determinado.
Genoma: todo el conjunto de órdenes genéticas que se encuentran en una célula. En los seres humanos, el genoma consta de 23 pares de cromosomas, que se encuentran en el núcleo, así como un pequeño cromosoma que se encuentra en las mitocondrias de las células. Cada conjunto de 23 cromosomas contiene aproximadamente 3.100.000.000 bases de secuencia de ADN.
Genotipo: colección de genes de una persona. El término también puede hacer referencia a los dos alelos heredados de un gen determinado.
Heterocigótico: hace referencia al hecho de haber heredado diferentes formas de un gen determinado de cada padre. Un genotipo heterocigótico es diferente a un genotipo homocigótico, en que una persona hereda formas idénticas de un gen determinado de cada padre.
Homocigótico: condición genética en que una persona hereda los mismos alelos de un gen determinado de ambos padres.
Mutación: un cambio en la secuencia de ADN que puede estar provocado por radiación, infecciones víricas, exposición a productos químicos o errores durante la división celular. Las mutaciones de la estirpe germinal se producen en huevos y esperma, y pueden transmitirse a la descendencia; las mutaciones somáticas no se transmiten a medida puesto que se producen en las células del cuerpo.
Penetrancia: probabilidad de que se exprese un gen o rasgo genético. Penetrancia completa significa que el gen o genes de un rasgo se ha expresado en toda la población que tienen los genes. Penetrancia incompleta significa que el rasgo genético se expresa solo en una parte de la población. El porcentaje de penetrancia también puede cambiar con el rango de edad de la población.
Portador: persona que lleva y es capaz de transmitir una mutación genética asociada a una enfermedad y puede presentar síntomas de la enfermedad o no. Los portadores se asocian con enfermedades heredadas como rasgos recesivos. Para presentar la enfermedad, una persona debe haber heredado alelos mutados de ambos progenitores. Una persona con un alelo normal y un alelo mutado no presenta la enfermedad. Dos portadores pueden tener descendencia con la enfermedad.
Los pacientes con dos copias de la mutación genética más frecuente, C282Y, por lo general, pero no siempre, presentan síntomas y signos de HH relacionados con la penetrancia. La mayoría de los pacientes sintomáticos que solicitan atención médica son adultos mayores de 40 años. Los hombres pueden verse afectados antes que las mujeres porque la menstruación reduce la acumulación de hierro y retrasa la aparición de los síntomas hasta después de la menopausia12,13. El inicio de los síntomas también se puede retrasar en personas que donan sangre de forma habitual, pues ello elimina el exceso de hierro.
En las primeras etapas, un paciente con HH puede permanecer asintomático. Se desarrollan algunas manifestaciones clínicas inespecíficas, ya que el hierro se acumula en tejidos y órganos. Entre los ejemplos se pueden citar debilidad, dolor abdominal y pérdida de peso involuntaria. Algunos de los trastornos más graves asociados con la enfermedad avanzada son cirrosis, diabetes mellitus, cardiomiopatía, artritis e hipogonadismo.
Una presentación tardía y poco frecuente es la “diabetes bronceada”, que se caracteriza por la tríada de cirrosis, diabetes e hiperpigmentación de la piel, lo que confiere a la piel un color bronceado7. Esta presentación se ha vuelto menos frecuente porque a la mayoría de los pacientes hoy día se les diagnostica y trata antes que desarrollen complicaciones3,12.
Alguien con HH puede donar sangre de una flebotomía terapéutica porque la HH no es una hemopatía: los eritrocitos y otros glóbulos sanguíneos no se ven afectados y la HH no se transmite por la sangre. El paciente puede programar flebotomías terapéuticas en hospitales, clínicas o unidades móviles o centros comunitarios y hospitalarios de donación de sangre. Aconseje al paciente para que elija un centro o lugar que pueda realizar flebotomías con la frecuencia prescrita por el médico.
Antes y después de una flebotomía terapéutica, el paciente debe beber agua, leche o zumo de frutas para tratar de que aumente el flujo sanguíneo durante la flebotomía y reponer los líquidos después. Aconseje al paciente que evite la actividad física vigorosa durante las 24 horas después de un tratamiento.
El paciente continuará precisando flebotomías terapéuticas de por vida. Haga hincapié en la importancia de controlar la acumulación de hierro como lo indique el médico y de realizar flebotomías según lo prescrito para evitar complicaciones y una muerte prematura.
El Instituto de Trastornos del Hierro dispone de recursos para los pacientes en relación con los programas de donantes con hemocromatosis en www.irondisorders.org
Se le ha diagnosticado hemocromatosis, una enfermedad que provoca que su cuerpo absorba una cantidad excesiva de hierro. La afección generalmente es hereditaria y se transmite a través de los genes. Sus familiares consanguíneos necesitan hablar con su médico para hacerse un sencillo análisis de sangre y así saber si tienen la misma afección. Los CDC ponen a su disposición una carta que puede informarles sobre la afección (v. www.cdc.gov/ncbddd/hemochromatosis/training/pdf/hemo_family_letter.pdf). Entregue esta carta a sus familiares consanguíneos: hermanas, hermanos, madre, padre, hijos adultos (informe al médico de sus hijos pequeños) y los abuelos por ambas partes de la familia. Los miembros de la familia deben entender que, si han heredado los genes que originan la enfermedad, el hierro puede acumularse en el corazón, las articulaciones, el hígado o el páncreas, y causar una lesión permanente, incluso si en ese momento se encuentran bien. El diagnóstico y tratamiento precoces pueden prevenir complicaciones.
Para controlar su enfermedad y prevenir complicaciones, siga estas indicaciones:
- •
Controle los niveles de hierro y reciba el tratamiento con flebotomía que le prescriba su médico. La hemocromatosis no puede tratarse solamente con cambios en la dieta.
- •
No tome pastillas de hierro ni ningún suplemento nutricional o multivitamina que contenga hierro. Lea atentamente las etiquetas de los productos.
- •
No tome vitaminas ni suplementos que contengan más de 500 mg de vitamina C al día, ya que la vitamina C aumenta la cantidad de hierro que su cuerpo absorbe. Se recomienda el consumo de alimentos que contengan vitamina C.
- •
No coma pescado ni marisco crudo, ya que puede contener gérmenes que pueden ser perjudiciales para las personas con sobrecarga férrica. Sin embargo, el consumo de pescado y marisco bien cocinado está bien ya que la cocción destruye los gérmenes.
- •
Si decide beber alcohol, beba muy poco (no más de 1 copa/día en el caso de las mujeres o 2 copas/día en el caso de los hombres). Si tiene problemas hepáticos, evite el alcohol por completo.
- •
Para obtener más información y recursos, visite el sitio web de los CDC en www.cdc.gov/ncbddd/hemochromatosis/index.html
Los síntomas y signos de HH reflejan la acumulación de hierro en diversos tejidos y órganos3:
- •
La acumulación de hierro en el hígado puede causar hepatomegalia, fibrosis y cirrosis, que puede producir insuficiencia hepática, carcinoma hepatocelular y muerte.
- •
La acumulación de hierro en el páncreas puede causar diabetes mellitus, que está presente en aproximadamente el 50% de los pacientes con HH sintomáticos12.
- •
El dolor en las articulaciones no se entiende completamente, pero puede estar relacionado con el exceso de hierro que produce cristales de calcio en los tejidos de las articulaciones, lo que provoca dolor en las articulaciones y deformidad, especialmente en las articulaciones metacarpofalángicas.
- •
La acumulación de hierro en los tejidos del corazón puede causar miocardiopatía dilatada, trastornos de la conducción e insuficiencia cardíaca.
- •
La acumulación de hierro en las células de la hipófisis puede provocar un descenso de los niveles de hormonas sexuales, lo que genera hipogonadismo e impotencia en hombres, y amenorrea en mujeres.
- •
La acumulación de hierro en la glándula tiroides puede reducir los niveles de hormona tiroidea y causar hipotiroidismo.
- •
La acumulación de hierro y melanina en la piel puede causar hiperpigmentación y generar un tono de piel bronceada o de color bronce.
Debilidad, cansancio y letargo están presentes en aproximadamente el 75% de las personas sintomáticas en el momento del diagnóstico12. Dado que los efectos sobre los mecanismos fisiológicos normales son tan importantes, el diagnóstico precoz es fundamental para evitar el desarrollo de problemas de salud graves.
Pruebas de diagnósticoEl diagnóstico de la HH depende de la existencia de un aumento de las reservas de hierro con síntomas o sin ellos. Inicialmente, se obtienen los niveles de ferritina sérica y de saturación de transferrina. La ferritina es una proteína que almacena hierro en el cuerpo. La producción de ferritina sérica aumenta cuando se absorbe el exceso de hierro. Los niveles de ferritina sérica son el indicador más fiable de las reservas de hierro de todo el cuerpo14.
La transferrina es una proteína de transporte que recoge hierro absorbido por los intestinos y lo transporta dentro del cuerpo. Cuando la absorción de hierro es anormalmente elevada, las proteínas de transferrina se saturan con hierro, por lo que un nivel elevado de saturación de transferrina indica aumento de la absorción de hierro14.
Si cualquiera de estos valores es anormal (saturación de transferrina ≥ 45% o ferritina por encima del límite superior de la normalidad, >300 ng/ml en hombres), entonces se debe llevar a cabo el análisis de la mutación del gen HFE para confirmar el diagnóstico2–14.
- •
La Asociación Americana de Enfermeras (ANA, American Nurses Association) ha definido las competencias para todos los niveles de enfermería en la segunda edición de Essentials of Genetic and Genomic Nursing: Competencies, Curricula Guidelines, and Outcome Indicators (Fundamentos de genética y genómica para enfermería: competencias, guías de planes de estudio e indicadores de resultados). https://www.genome.gov/Pages/Careers/HealthProfessionalEducation/geneticscompetency.pdf
- •
La ANA y la Sociedad Internacional de Enfermeras en Genética (ISONG, International Society of Nurses in Genetics) publicaron Essential Genetic and Genomic Competencies for Nurses with Graduate Degrees (Competencias esenciales en genética y genómica para enfermeras con títulos de posgrado) para definir un mayor nivel de experiencia para las enfermeras con títulos de máster y doctorado. www.genome.gov/Pages/Health/HealthCareProvidersInfo/Grad_Gen_Comp.pdf
- •
ISONG es una organización global de la especialidad de enfermería dedicado a cuidados, educación, investigación y becas en genómica. Seminarios y otras oportunidades educativas están disponibles en www.isong.org/index.php
- •
El Centro de Competencias en Genética/Genómica (The Genetics/Genomics Competency Center) ofrece recursos en genética y genómica para su uso en el aula o en la práctica. Visite http://g-2-c-2.org
- •
La Coalición Nacional de Educación Profesional en Salud en Genética (NCHPEG, National Coalition for Health Professional Education in Genetics) es un recurso interdisciplinario para promover la colaboración entre un grupo diverso de personas influyentes. La ANA se unió a la Asociación Médica de Estados Unidos (American Medical Association) y al Instituto Nacional de Investigación del Genoma Humano (National Human Genome Research Institute) para crear la NCHPEG con el objetivo de promover la educación y el acceso a la información sobre los avances en genética humana. Más información en www.nchpeg.org
- •
El Audioglosario de Términos Genéticos del Instituto Nacional de Investigación del Genoma Humano (National Human Genome Research Institute) es un recurso gratuito, que puede descargarse en dispositivos móviles o el ordenador. Los términos genéticos y genómicos se definen mediante texto y versiones de audio. Para obtener más información, consulte www.genome.gov/glossary
- •
Telling Stories (Contar historias) es un recurso educativo multimedia en la web que utiliza historias de la vida real del público y los profesionales para promover el conocimiento y la comprensión de la genética y la forma en que afecta a la vida de las personas. La colección de 100 historias de personas, pacientes, cuidadores y profesionales en formatos de texto y vídeo se organizan en esquemas didácticos para enfermeras, comadronas, estudiantes de medicina y médicos de atención primaria. Visite www.tellingstories.nhs.uk
En el pasado era necesaria una biopsia de hígado para establecer el diagnóstico definitivo de HH. Sin embargo, el análisis de la mutación del gen HFE ha reducido la necesidad de biopsia hepática aunque puede estar indicada en ciertos pacientes para determinar la gravedad de la enfermedad o de una enfermedad hepática subyacente, como la infección por el virus de la hepatitis C3–13.
Las pruebas genéticas también pueden identificar a los portadores, lo que puede ser de utilidad para las parejas que planean tener descendencia. Uno de cada dos individuos portadores asintomáticos podría aportar una copia mutada del gen a un hijo que podría desarrollar la enfermedad. Es importante la intervención temprana para prevenir daños en los órganos de los individuos afectados, por lo que la identificación de un hijo en situación de riesgo es importante para evitar complicaciones2,9.
Tratamiento de por vidaLos pacientes con sobrecarga férrica sintomáticos son tratados con flebotomía para eliminar el exceso de hierro. Después de corregir la sobrecarga férrica, se realiza un seguimiento de los pacientes y se los trata con flebotomía, según sea necesario.
Inicialmente, durante la fase de inducción, se extrae una cantidad fija de sangre dos veces por semana, durante una semana o un mes, dependiendo de la situación clínica del paciente. La duración de este agresivo tratamiento depende del nivel de ferritina del paciente, el sexo y otros factores del paciente15.
Cuando los niveles de ferritina se sitúan por debajo de un nivel objetivo, como 500 ng/ml, la flebotomía se puede realizar con menos frecuencia, por ejemplo, cada mes. Durante esta fase de transición, el objetivo es reducir los niveles de ferritina de manera gradual a niveles normales. El tratamiento entra entonces en la fase de mantenimiento, en la cual la flebotomía se lleva a cabo según sea necesario para mantener niveles de hierro seguros15.
El Instituto de Trastornos del Hierro (The Iron Disorders Institute) recomienda alcanzar un nivel de ferritina sérica por debajo de 50 ng/ml, al menos una vez durante la fase de transición del tratamiento, y después adaptar el tratamiento para mantener los niveles en el intervalo de 25 a 75 ng/ml15. Llegados a este punto, el paciente quizá requiera flebotomía solo unas pocas veces al año15,16.
Los niveles de hematócrito/hemoglobina del paciente se revisan antes de cada flebotomía. El comité asesor del Instituto de Trastornos del Hierro no recomienda la flebotomía (con algunas excepciones) si la hemoglobina está por debajo de 12,5 g/dl para prevenir la anemia ferropénica15.
El inicio de los síntomas también se puede retrasar en personas que donan sangre de forma habitual, pues ello elimina el exceso de hierro.
Otro posible tratamiento es la terapia de quelación con deferoxamina o deferasirox. El beneficio de la terapia de quelación consiste en el hecho de que el fármaco se une fuertemente al hierro y lo elimina del cuerpo, con lo que se reducen las reservas de hierro. Sin embargo, este tratamiento se utiliza muy poco para tratar la HH ya que la flebotomía es más sencilla e igualmente eficaz12.
Pautas dietéticasA pesar de que la recomendación de seguir una dieta es una parte importante del plan de tratamiento de un paciente con HH, no es necesario que esta sea una dieta especial: una cantidad moderada de alimentos ricos en hierro no empeoran la enfermedad. Sin embargo, debe instruirse al paciente con el objetivo de que evite vitaminas o suplementos que contengan hierro o vitamina C, lo que puede aumentar la absorción de hierro. El alcohol con moderación es admisible a excepción de que al paciente se le haya diagnosticado enfermedad hepática. En ese caso, el paciente debe evitar el alcohol.
El paciente también debe evitar el marisco crudo, ya que puede contener bacterias que crecen bien en un entorno rico en hierro. Vibrio vulnificus, una bacteria que se encuentra en el marisco crudo y en las ostras, se desarrolla en la sangre y los órganos ricos en hierro, y puede ser mortal si se ingiere; se han comunicado muertes de personas con HH que ingirieron ostras crudas8,13,17.
Prevención de complicacionesEn los últimos años, el número de pacientes que desarrollan complicaciones graves relacionadas con la HH se ha reducido debido a algunos factores, como mayor atención a los resultados anómalos de análisis de sangre normales, pruebas más generalizadas de personas con un familiar diagnosticado de esta enfermedad e identificación de los pacientes con HH a una edad más temprana. Como resultado, la mayoría de las personas no presenta complicaciones en el momento del diagnóstico12.
Puesto que la mutación genética se transmite de ambos progenitores a un hijo, los médicos recomiendan que se hagan pruebas a los familiares de primer grado (padres, hermanos e hijos) de personas con HH. El objetivo de las pruebas es detectar HH antes de que se presenten los síntomas o las complicaciones. El hermano o hermana de una persona diagnosticada con HH tiene una probabilidad de cuatro de presentar el trastorno tras haber heredado dos copias de la mutación C282Y.
Idealmente, los familiares de primer grado deben realizarse pruebas entre los 18 y los 30 años, antes que haya daño tisular grave. La recomendación de la primera fase de detección es solicitar un análisis de sangre para determinar si existe saturación de transferrina y los niveles de ferritina sérica. Si los resultados indican un aumento de las reservas de hierro en consonancia con la HH, el médico recomendará proceder a la realización de pruebas genéticas para detectar la mutación del gen HFE2. Si una pareja tiene o planea tener hijos, también se recomienda que el cónyuge de la persona afectada realice las pruebas para determinar si lleva la mutación del gen HFE. En internet hay recursos de los CDC para los pacientes y sus familias en www.cdc.gov/ncbddd/hemochromatosis/materials.html
El estudio de caso continuóUna búsqueda en la bibliografía acerca de la HH por parte de la enfermera que atendía al Sr. W. reveló que entre sus factores de riesgo se contaban la edad, el sexo, el origen étnico, los antecedentes familiares y el hecho de que nunca había donado sangre. El análisis de los datos de la evaluación física indicaba que sus síntomas poco precisos de cansancio, dolores musculares, malestar abdominal y dolor de rodilla respondían a la fisiopatología de la HH. Se estableció un plan de tratamiento médico que incluía flebotomía con el control de los valores de la ferritina sérica, la hemoglobina y el hematócrito.
La educación en el momento del alta consistió en instrucciones pre y posflebotomía, así como una conversación sobre modificaciones en la dieta; por ejemplo, educación para evitar suplementos de hierro o multivitaminas con hierro. También era importante la necesidad de que el paciente informara a sus familiares consanguíneos sobre el riesgo de padecer este trastorno (para obtener más detalles, v. Educación sanitaria del paciente: comparta esta información con los pacientes y sus familias, y sugerencias para flebotomía).
El compromiso de la enfermera de aprender más sobre el diagnóstico del señor W. está de acuerdo con la responsabilidad de enfermería de ofrecer cuidados competentes a los pacientes con trastornos genéticos18 (v. Recursos para mantenerse al día). Al lograr y mantener la competencia en el cuidado de pacientes con trastornos genéticos, las enfermeras contribuyen a integrar los avances científicos de la genética y la genómica en la práctica clínica. ■
Los autores y los editores declaran no tener ningún conflicto de intereses económico o de otro tipo relacionado con este artículo.