


MADRE DE TRES adolescentes, la señora S., de 49 años, fue al médico con fasciculaciones y debilidad en ambas piernas, dificultad para tragar y fatiga, que se había ido agravando durante las últimas semanas. Mientras paseaba una mañana, tropezó y cayó al suelo. Se hizo un pequeño corte en la pierna, por lo que se dirigió a un centro sanitario. Su marido explicó que durante los últimos meses había presentado episodios de habla farfullante. Estaba despierta y orientada, y sus signos vitales estaban dentro de los límites normales.
Después de una exploración, el médico la remitió al neurólogo, quien le mandó que se hiciera una resonancia magnética (RM), un electromiograma y una analítica de sangre completa. Después de varias visitas para descartar otras causas, la Sra. S. recibió el demoledor diagnóstico de esclerosis lateral amiotrófica (ELA).
La ELA, conocida también como enfermedad de Lou Gehrig, por el nombre del famoso jugador de béisbol que sufrió esta enfermedad, es una de las enfermedades neuromusculares más frecuentes en el mundo. Esta enfermedad neuromuscular rápidamente progresiva y mortal consiste en la degeneración y muerte de las motoneuronas superiores e inferiores1. Este artículo aborda el diagnóstico y tratamiento de la ELA y cómo las enfermeras pueden ayudar a sus pacientes a lidiar con el difícil diagnóstico y a encontrar los recursos que estos y sus familias necesitan.
¿A quién afecta?Se estima que entre 20.000 y 30.000 estadounidenses viven con ELA y se diagnostican unos 5.000 nuevos casos cada año en Estados Unidos2. Esta enfermedad afecta a personas de cualquier origen étnico, socioeconómico y racial2,3. La ELA afecta con más frecuencia a personas entre 40 y 60 años, pero también puede desarrollarse en personas más jóvenes y mayores. La incidencia es ligeramente mayor en hombres que en mujeres3.
La media de supervivencia después del diagnóstico es de 3 a 5 años, pero la enfermedad progresa más lentamente en algunos casos3. Los informes de la ALS Association indican que alrededor del 30% de las personas con ELA vive 5 años, del 10 al 20% sobrevive más de 10 años y el 5% vive 20 años4. Sin embargo, la supervivencia incluso a más largo plazo es posible si la enfermedad se manifiesta a una edad más temprana. Un ejemplo de ello es el famoso físico teórico Stephen Hawking. Al Dr. Hawking se le diagnosticó la enfermedad a los 21 años y lleva viviendo con ella más de 50 años. Aunque está confinado a una silla de ruedas y utiliza un sintetizador de voz para comunicarse, expuso su propia teoría de la relatividad y la mecánica cuántica, trabajó durante más de 30 años como catedrático de matemáticas y fundó el Stephen Hawking Centre for Theoretical Cosmology (Cambridge, Reino Unido), padeciendo ELA5.
Aparte del diagnóstico a una edad más temprana, otras variables para la supervivencia a más largo plazo son el sexo masculino y la aparición de la enfermedad en las extremidades frente al inicio bulbar6 (v. el cuadro Comprender la fisiopatología).
Etiología desconcertanteHasta la fecha, no se ha identificado ninguna causa única de la ELA. En el 90-95% de casos de ELA, al parecer la enfermedad ocurre espontáneamente2. Los pacientes con esta forma esporádica de la enfermedad no tienen antecedentes familiares de ELA y sus familiares no corren mayor riesgo de presentar la enfermedad2.
Aproximadamente, el 5-10% de todos los casos de ELA se heredan. La ELA familiar (ELAF) generalmente se debe a un patrón de herencia autosómica dominante que solamente necesita que uno de los progenitores porte el gen causante de la enfermedad. La edad de inicio de la ELAF es de unos 10 años antes que la media de inicio de ELA y se asocia con una supervivencia más corta7.
Un tercio, más o menos, de todos los casos familiares se debe a un defecto en un gen conocido como cromosoma 9 marco abierto de lectura 72 o C9orf72, un gen cuya función todavía es confusa. Esta mutación en el gen también está implicada en un pequeño porcentaje de casos no familiares. Otro 20% de los casos familiares se debe a mutaciones en el gen que codifica la enzima superóxido dismutasa de tipo 1 (SOD1) que contiene cobre y cinc2. Se dispone de pruebas genéticas para cualquier persona con indicios de ELA y antecedentes familiares de ELA. Aunque las pruebas genéticas habitualmente no forman parte de la evaluación diagnóstica de la ELA, pueden ayudar a determinar la causa de la ELAF de una familia. La prueba es más útil cuando a una persona en la familia ya se le ha diagnosticado la enfermedad. Aproximadamente, el 50% de las personas con ELAF tendrá una mutación identificada. Si se ha identificado una mutación, se puede hacer la prueba a los familiares biológicos asintomáticos para determinar si heredaron la mutación genética. Esto se conoce como pruebas predictivas8.
Otras causas posibles objeto de investigación son el tabaquismo, una infección viral, enfermedades autoinmunitarias y exposición ambiental a toxinas4. Se realizan estudios en personal militar que fue enviado a la guerra del Golfo de 1991. Un estudio reciente demostró que los veteranos eran más propensos a desarrollar ELA en comparación con el personal militar que no estuvo en la región2,4.
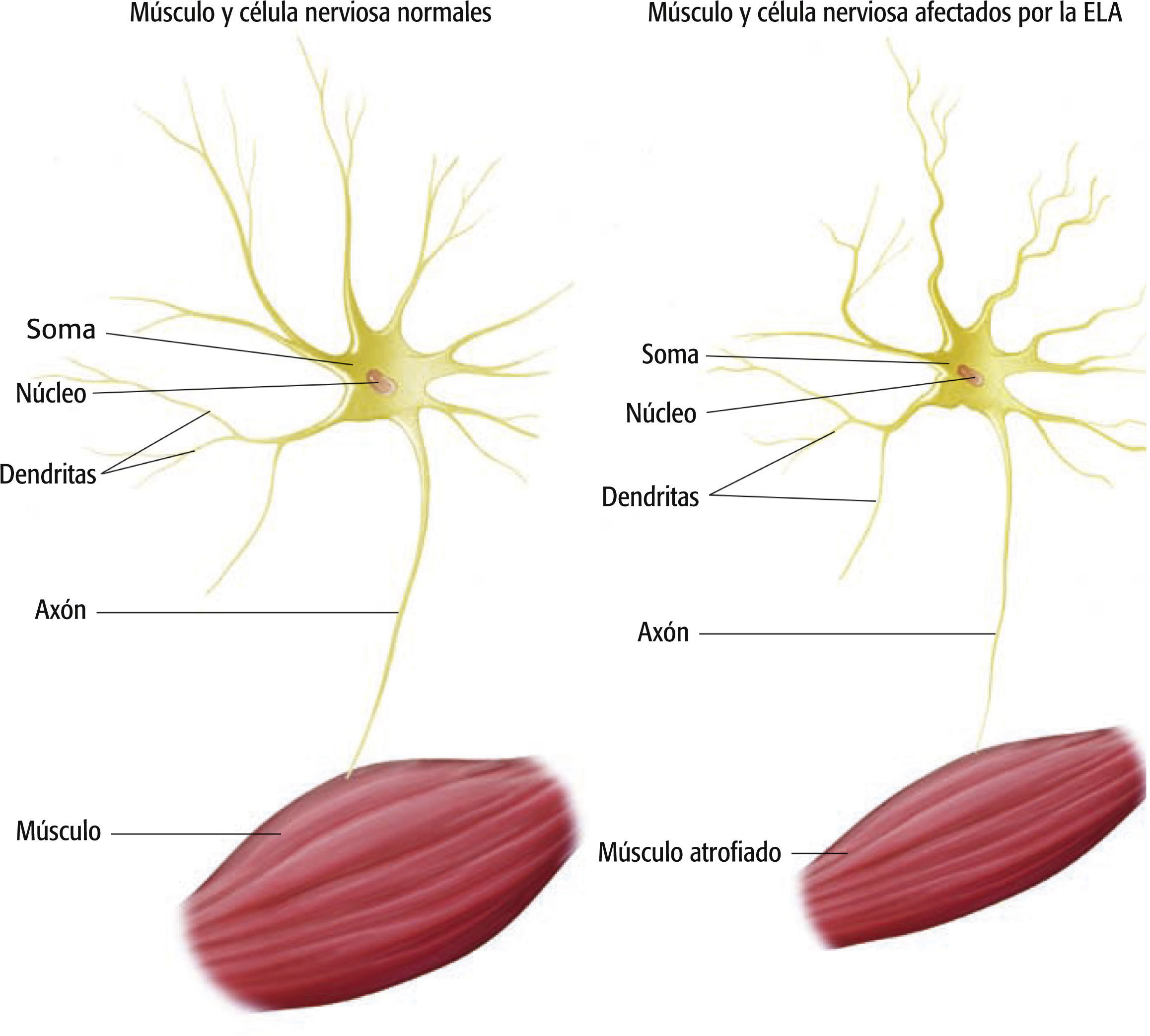
Comprender la fisiopatología19
La ELA conduce a la degeneración progresiva de las motoneuronas que inervan los músculos voluntarios. Se caracteriza por el deterioro de las neuronas piramidales en la corteza motora (motoneuronas superiores) y de las motoneuronas en el tronco encefálico y la médula central (motoneuronas inferiores). Esta degeneración progresiva de las motoneuronas produce finalmente la muerte. Cuando las motoneuronas mueren, el cerebro pierde la capacidad de iniciar y controlar el movimiento de los músculos y los músculos empiezan a atrofiarse. A esto se le denomina amiotrofia. La pérdida de fibras nerviosas en las columnas laterales de la médula espinal provoca rigidez o esclerosis en el tejido del sistema nervioso central; de ahí el término esclerosis lateral. Sin embargo, no quedan afectados todo el sistema sensorial y los métodos de regulación del control y coordinación de los movimientos. Las neuronas del movimiento ocular, las neuronas parasimpáticas de la médula espinal del sacro y el intelecto permanecen intactos.
Los signos y síntomas iniciales de ELA implican una progresión lenta y crónica que varía entre los afectados9. En el 80% de los pacientes se presenta debilidad asimétrica en un miembro mientras que el 20% tiene inicio bulbar, que se presenta con disartria o disfagia10.
Las primeras manifestaciones de la enfermedad dependen de la localización de las neuronas afectadas y pueden ser tan leves que la persona no las reconozca como un problema11. La persona probablemente no buscará tratamiento hasta que sean evidentes la atrofia muscular y la pérdida de peso9.
La característica clínica distintiva de la ELA es la combinación de signos y síntomas de las motoneuronas superiores e inferiores. Los hallazgos de debilidad, hiperreflexia y espasticidad de la motoneurona superior se deben a la degeneración de las motoneuronas frontales. Los hallazgos de debilidad, atrofia o amiotrofia y fasciculaciones de la motoneurona inferior son consecuencia directa de la degeneración de motoneuronas inferiores en el tronco encefálico y la médula espinal10.
Los signos y síntomas bulbares incluyen disfagia, que aumenta el riesgo de aspiración, y disartria9,11. Generalmente se respeta la función de la vejiga y el intestino dado que los nervios raquídeos, que controlan estos músculos, no están afectados11,12. Aunque aproximadamente el 15% de los pacientes con ELA pueden desarrollar demencia frontotemporal, la función cognitiva generalmente no está afectada en la mayoría de pacientes con ELA12.
Casi todos los pacientes desarrollan signos y síntomas de insuficiencia respiratoria que requieren ventilación mecánica. Las enfermeras deben asegurarse de tratar con los pacientes y sus familias los riesgos y beneficios de la ventilación invasiva mucho tiempo antes de tomar la decisión. Aproximadamente, el 5-10% de los pacientes elige la opción de una traqueostomía con ventilación mecánica permanente cuando la insuficiencia respiratoria se vuelve grave. La causa más frecuente de muerte en casos de ELA es la insuficiencia respiratoria neuromuscular progresiva. Otras causas de muerte son la aspiración y la infección10.
Diagnosticar la ELAEstablecer un diagnóstico de ELA es una tarea difícil que puede llevar mucho tiempo porque no se puede confirmar con una única prueba2,12. Más bien, el diagnóstico se basa generalmente en los signos y síntomas que se presentan y en la exclusión de otras causas posibles12. Las técnicas de neuroimagen, que incluyen TC y RM cerebrales, se utilizan para descartar trastornos que pueden parecerse a la ELA, como la esclerosis múltiple o un tumor cerebral9,12. Los estudios electrodiagnósticos son útiles en la evaluación de la debilidad, la atrofia muscular y los síntomas sensoriales. Por lo general, en la ELA revelan características combinadas de desnervación aguda y crónica13. Estudios de conducción sensitiva nerviosa pueden determinar la capacidad de los nervios para enviar señales eléctricas a pesar de que los resultados de estos estudios son a menudo normales en pacientes con ELA13. Sin embargo, en casos de músculos atróficos y desnervados graves, los resultados de los estudios de conducción pueden mostrar reducción de la amplitud2,9.
Orientaciones para la investigación sobre la ELA
Entre los temas actuales de investigación sobre la ELA se encuentran los siguientes:
- •
Comprensión de los mecanismos que activan selectivamente las motoneuronas para que degeneren en la ELA y desarrollo de estrategias efectivas para detener los procesos que conducen a la muerte celular.
- •
Desarrollo de biomarcadores para la ELA que podrían ayudar a establecer el diagnóstico, que sirvan como marcadores de progresión de la enfermedad o se correlacionen con objetivos terapéuticos.
- •
Pruebas con compuestos semejantes a fármacos, enfoques de terapia génica, anticuerpos y terapias basadas en células14.
- •
Explorar el efecto del trasplante de células para tratar los signos y síntomas20,21.
- •
Estudiar la posible asociación entre tabaquismo y la ELA22.
Las pruebas de laboratorio generalmente incluyen electroforesis de proteína en orina y suero, estudios de tiroides y pruebas de detección de metales pesados en orina de 24 horas4,9. Entre otros estudios diagnósticos se encuentran hemograma completo con fórmula leucocitaria, velocidad de sedimentación globular, proteína C reactiva, vitamina B12 y niveles de folato9.
Elección del tratamientoLa ELA es progresiva e incurable, y los pacientes necesitan cuidados multidisciplinarios. Hasta la fecha, el riluzol es el único tratamiento que se ha demostrado que aumenta el tiempo de supervivencia14. Se piensa que el riluzol, un miembro de la clase del benzotiazol, reduce la excitotoxicidad provocada por el glutamato15,16. El riluzol puede alargar varios meses el tiempo de supervivencia en algunos pacientes y también puede prolongar el tiempo antes que el paciente necesite ventilación mecánica. Sin embargo, este medicamento no puede revertir los daños ya ocasionados. Entre las reacciones adversas observadas con más frecuencia pueden citarse astenia, náuseas, vómitos, diarrea, dolor abdominal, anorexia y mareos. Puesto que cerca del 10% de las personas que toman este medicamento presenta lesión hepática, la función hepática debe controlarse con frecuencia16.
La mayoría de los demás tratamientos de la ELA se utilizan para mejorar la calidad de vida de los pacientes y aliviar los signos y síntomas. El objetivo de este tratamiento de apoyo es ayudar a los pacientes a permanecer tan móvil, independiente y cómodo como sea posible. Puede indicarse farmacoterapia para el tratamiento de la disnea, espasmos musculares, espasticidad, sialorrea (flujo excesivo de saliva), fatiga, dolor, depresión, disfagia, estreñimiento y problemas de sueño.
Para los pacientes con ELA, la fisioterapia y la terapia ocupacional desempeñan un importante papel en el retraso de la pérdida de fuerza, mantenimiento de la resistencia, prevención de las complicaciones, reducción o control del dolor, y promoción de la independencia funcional. Los pacientes pueden beneficiarse de ejercicios aeróbicos de bajo impacto, como nadar, caminar y montar en bicicleta estática, que pueden ayudar a mejorar la salud cardiovascular y a combatir el cansancio y la depresión. El objetivo es proporcionar estrategias y herramientas a los pacientes que les ayuden a mantener la independencia y a garantizarles seguridad mientras realizan las actividades de la vida diaria. Los logopedas pueden ayudar a los pacientes que sufran disfagia y disartria17.
Para mantener la nutrición, anime a los pacientes a realizar varias comidas pequeñas al día. Las comidas deben proporcionar fibra, líquidos y calorías en alimentos que sean fáciles de tragar. Dado que la asfixia por exceso de saliva es una preocupación, siempre hay que mantener cerca los aparatos de aspiración18.
Los pacientes pueden necesitar que se les coloque una sonda de gastrostomía percutánea para el soporte nutricional6. Para el soporte pulmonar puede utilizarse ventilación con presión positiva no invasiva, traqueostomía y ventilación mecánica6.
Deberían proporcionarse cuidados paliativos desde el momento del diagnóstico y durante todo el proceso de pérdida para brindar apoyo al paciente con ELA, su familia y el equipo de cuidados. Los cuidados paliativos no se limitan a los cuidados al final de la vida sino más bien se centran en aliviar el sufrimiento a lo largo de cada etapa de la enfermedad. Entre los objetivos de los cuidados paliativos pueden citarse el establecimiento de objetivos de cuidados que son consecuentes con los valores físicos, psicosociales, emocionales y espirituales del paciente. Cuando el pronóstico vital del paciente es de 6 meses o menos, o cuando los objetivos buscan exclusivamente la comodidad, deberá buscarse un centro de cuidados paliativos6.
Investigación en cursoEl National Institute of Neurological Disorders and Stroke, que forma parte de los National Institutes of Health, es un destacado defensor de la investigación biomédica sobre la ELA. Los objetivos de esta investigación son encontrar la causa de la ELA, llegar a comprender los mecanismos implicados en la progresión de la enfermedad y desarrollar tratamientos efectivos. Los investigadores son optimistas respecto al hecho de que estos estudios de investigación clínica finalmente darán paso a tratamientos más efectivos para la ELA (v. el cuadro Orientaciones para la investigación sobre la ELA.)
De vuelta a la paciente…El médico de la señora S. le recetó riluzol oral y la remitió a un nutricionista y un fisioterapeuta. Se le programó una visita de seguimiento al cabo de 2 semanas. La señora S. y su familia necesitarán amplia educación sobre la enfermedad. Además de ofrecerle apoyo emocional y físico, la enfermera informa a la paciente y a su familia sobre los recursos disponibles en la población y le entrega materiales educativos.
Educación al pacienteCada paciente con ELA se ve afectado de forma diferente, por lo que hay que adaptar la educación al paciente y las intervenciones posteriores para satisfacer las necesidades específicas del paciente. Las enfermeras necesitan enseñar a los pacientes y a sus cuidadores la importancia de seguir las diferentes pautas de tratamiento prescritas y notificar al médico si debe haber cambios. Entre los temas que deben enseñarse pueden citarse:
- •
Supervisar los cambios en las vías respiratorias, la deglución o el habla.
- •
Ofrecer cuidados de la piel y prevenir las úlceras por presión.
- •
Intentar que la dieta del paciente esté bien equilibrada.
- •
Seguir las pautas de fisioterapia y terapia ocupacional.
- •
Controlar las alteraciones en los patrones de evacuación.
- •
Implementar estrategias de prevención de caídas, como la eliminación de posibles peligros de tropiezo, la instalación de barandillas y una silla para la ducha, y utilizar dispositivos de ayuda, como un andador o una silla de ruedas cuando sea necesario.
Ofrecer apoyo emocional y educación a los pacientes y sus familias durante el curso de la enfermedad son intervenciones de enfermería clave. Se recomiendan comunicación y escucha activa. Anime a los pacientes y a sus familias a que utilicen recursos que les ayudarán cuando se tengan que enfrentar a los desafíos de la ELA (v. el cuadro Recursos para pacientes y familias). ■