




A PESAR DE QUE CASI todo el mundo ha oído hablar de la distrofia muscular, todavía muchas enfermeras no están familiarizadas con la distrofia muscular de la cintura escapular y pélvica (DMC). Con el caso clínico que se presenta en este artículo, las enfermeras aprenderán a evaluar y cuidar a los pacientes con esta enfermedad hereditaria, a instruirlos en el cuidado personal y a familiarizarlos con los recursos de apoyo a los pacientes y a sus familias. Keith Brazzo, uno de los autores de este artículo, tiene una hija con esta enfermedad y escribe desde la experiencia personal.
Caso clínicoA ⿿Julie⿿ se le diagnosticó DMC de tipo 21 (DMC21) por primera vez a los 2 años. Los primeros síntomas y signos consistieron en marcha de pato, debilidad en la cadera, cinturas escapulares y tronco, deambulación levemente retrasada, hipertrofia de la pantorrilla y dificultad para levantarse de una posición sentada o del decúbito prono.
Tras numerosas evaluaciones de un traumatólogo pediátrico y varios especialistas neuromusculares, se realizó un análisis de sangre que reveló un nivel de creatina cinasa (CK) sérica superior a 15.000 U/l (valores normales: 26-140 U/l)1. Más adelante se le realizaron pruebas genéticas con un perfil de DMC, que confirmaron una mutación del gen de la proteína relacionada con fukutina, en consonancia con la DMC212.
Desde ese momento, Julie, que ahora tiene 9 años, ha recibido tratamientos paliativos, entre ellos fisioterapia y terapia ocupacional, terapia acuática y paseos a caballo para actuar sobre la musculatura lumbopélvica y de la cintura. Anualmente, el cardiólogo pediátrico le realiza una evaluación diagnóstica para controlar la función cardiovascular. Además, recibe suplementos, entre los cuales se encuentran la coenzima Q-10 y l-glutamina para estimular la función muscular y aumentar los niveles de energía3⿿5.
DMC: una forma de distrofia muscularLa distrofia muscular es un grupo de trastornos que generan debilidad y atrofia muscular. Las tres formas más frecuentes son las distrofinopatías (distrofia muscular de Duchenne y de Becker), distrofia miotónica y distrofia muscular facioescapulohumeral6. En 1954, Walton y Nattrass propusieron que la DMC se considerara un fenotipo clínico independiente para diferenciarlo de los otros tipos de distrofia muscular7,8. Actualmente, la DMC es la cuarta causa genética más frecuente de debilidad muscular6.
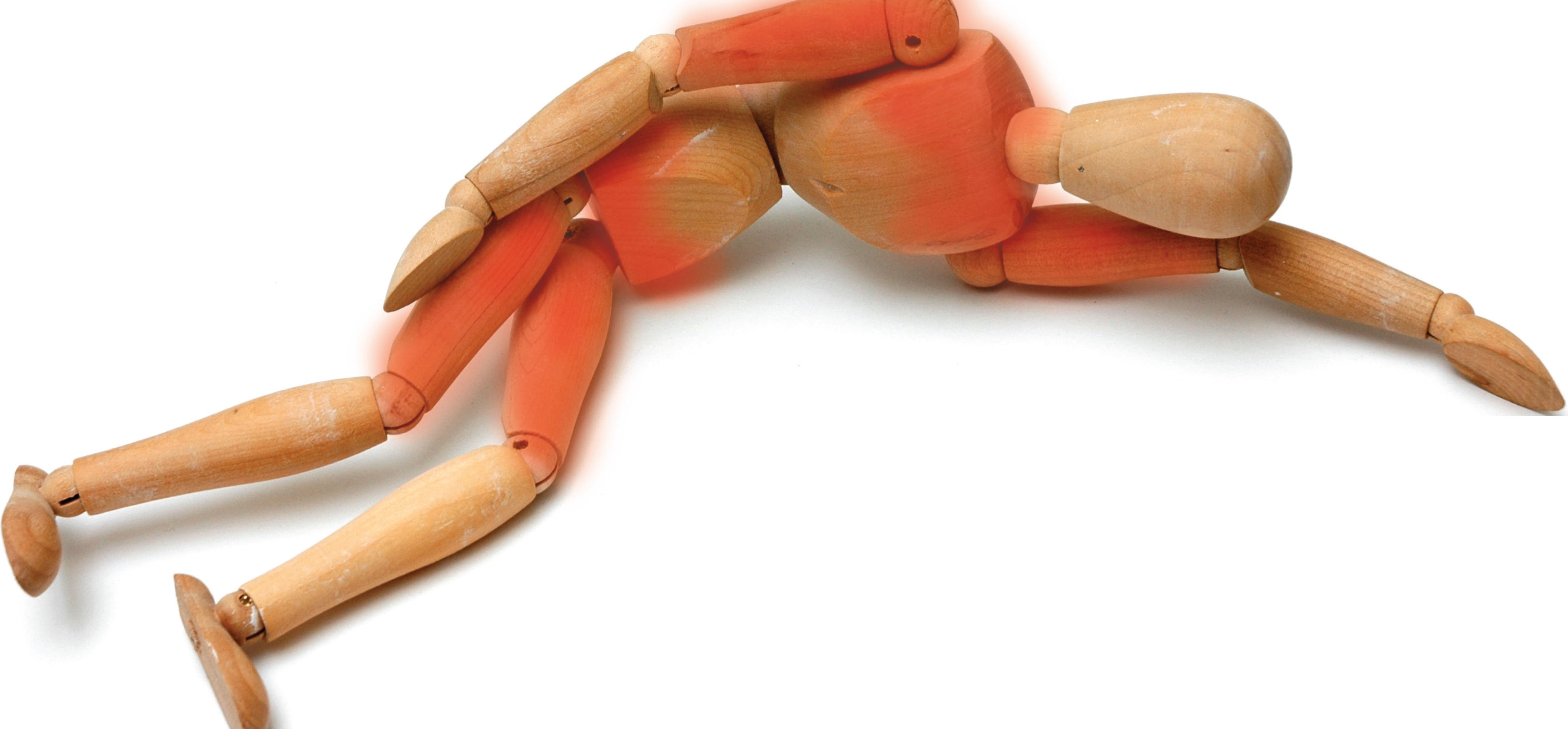
La DMC es una miopatía progresivamente debilitante que se caracteriza por debilidad y atrofia muscular principalmente en las zonas de la cintura escapular y la cintura pélvica. Esta extraña enfermedad genética generalmente comienza a principios de la adolescencia, pero puede aparecer en cualquier momento desde la primera infancia hasta la edad adulta6. Cuando el niño con DMC llega a la adolescencia, puede sufrir complicaciones cardiovasculares, respiratorias y adicionales debido a la movilidad limitada. Algunos subtipos de DMC tienen unas características distintivas, como patrones de afectación muscular, anomalías cardíacas y afectación extramuscular. Las biopsias musculares pueden descartar la inflamación y distinguir entre las diferentes formas de distrofia muscular (v. el cuadro ¿A quiénes afecta?).
¿A quiénes afecta?
Se calcula que la prevalencia mundial de todas las formas de DMC fluctúa entre 1 de cada 14.500 y 1 de cada 123.000 personas10,17. La mayoría de las DMC se heredan como un rasgo autosómico recesivo (DMC2), con una prevalencia que varía entre 0,07 por 100.000 y 0,43 por 100.000 personas6. La frecuencia de algunas distrofias musculares varía en función de la etnia de la población estudiada16,18⿿20. Hombres y mujeres se ven afectados por igual.
Según la definición de la Muscular Dystrophy Association, las DMC son enfermedades genéticas degenerativas que afectan a los músculos estriados, causadas por una mutación en cualquiera de los 15 genes que afectan a las proteínas necesarias para llevar a cabo la función muscular (v. el cuadro Comprender la fisiopatología de la DMC). La DMC1 remite a subtipos genéticos con herencia autosómica dominante, mientras que DMC2 hace referencia a subtipos con herencia autosómica recesiva. La mayoría de los casos de DMC son de tipo 2. En la actualidad se han cartografiado 23 formas diferentes, que se han designado con las letras 2A a 2W, en regiones cromosómicas específicas y genes específicos6,9.
Los nombres descriptivos se basan en el producto proteínico anómalo identificado. La superposición clínica entre entidades presenta desafíos en el momento de establecer un diagnóstico clínico preciso10.
Síntomas y signosLos síntomas y signos iniciales de la DMC se caracterizan por una debilidad muscular proximal primaria progresiva con afectación de la cintura escapular y la cintura pélvica11. Cada paciente presenta un aspecto único y tiene características distintivas según el subtipo específico de DMC. Cuando la enfermedad se inicia en la infancia, tiende a afectar a la cintura pélvica, pero cuando se presenta en la edad adulta, es más probable que afecte tanto a la cintura escapular como a la cintura pélvica, e incluye debilidad proximal de las extremidades que limita gradualmente la movilidad6.
Al igual que todas las distrofias musculares, la DMC es progresiva, pero el ritmo de progresión varía según el subtipo de DMC. La DMC autosómica recesiva (DMC2A-S) con inicio en la infancia por lo general progresa más rápidamente, lo que provoca la pérdida de movilidad al final de la infancia o la adolescencia, y la muerte en la edad adulta temprana, aunque también se presentan casos menos graves. Las formas autosómicas dominantes (DMC1A-F), incluso aquellas con inicio en la infancia, son más benignas y los pacientes pueden tener una esperanza de vida normal12.
En muchas personas con DMC, el primer signo es la marcha de pato o marcha ⿿intranquila⿿ debido a la debilidad de los músculos de la cadera y las piernas. Estas personas pueden tener problemas para levantarse de una posición sentada. La debilidad en el área del hombro es un gran problema para los pacientes cuando levantan la mano por encima de la cabeza, mantienen los brazos extendidos o cargan objetos pesados. Peinarse o colocar objetos en un estante alto es difícil para los pacientes. A algunos les resulta agotador escribir en un teclado o comer por sí solos13.
El corazón de un paciente casi nunca se ve afectado; ello depende del subtipo de DMC6,14. Los problemas pueden ser de dos tipos: miocardiopatía y trastornos de conducción cardíaca, o arritmias. Haga un seguimiento del paciente para detectar síntomas y signos de afectación cardíaca.
En pacientes con subtipos como la DMC2I puede haber músculos débiles implicados en la respiración, por lo que pueden desarrollar insuficiencia respiratoria. En el caso de pacientes que pueden correr este riesgo, controle el estado pulmonar con regularidad porque con el tiempo puede deteriorarse cuando avance la debilidad muscular. Algunos pacientes pueden beneficiarse de la ventilación no invasiva6.
Los pacientes con DMC no suelen quejarse mucho de dolor, pero la movilidad limitada a veces provoca mialgia y artralgia. Intente que el paciente no se canse demasiado porque esto puede empeorar la debilidad y el dolor. El descanso y el sueño adecuados son fundamentales11,15.
Los músculos de la faringe y de la región palatina permanecen intactos en la mayoría de las personas con DMC, aunque puede haber excepciones según el subtipo genético. Comer también puede ser más difícil a causa de la debilidad del brazo. Se deben realizar estudios de deglución de aquellos pacientes que aspiran o sufren disfagia o dificultad para mantener una nutrición adecuada, o se los debe derivar a un gastroenterólogo. Cambiar la consistencia de los alimentos, utilizar la posición encogida del mentón o establecer la nutrición enteral puede ayudar a los pacientes afectados6.
Los pacientes con DMC pueden correr mayor riesgo de deformidades musculoesqueléticas, como escoliosis o cifosis. Cualquier paciente que tenga alguno de estos problemas debería consultar a un especialista en ortopedia para valorarlo y tratarlo6.
La mayoría de los pacientes con DMC tienen un nivel intelectual normal, pero aquellos con DMC2K presentan discapacidad intelectual y algunos de los que tienen DMC2N también pueden verse afectados6.
Diagnosticar la DMCEn general, los niveles de CK sérica están ligeramente elevados en la DMC. Sin embargo, pueden ser muy altos en algunos subtipos de DMC6. Las elevaciones de CK sérica son una medida fiable de enfermedades óseas y musculares inflamatorias1. La electromiografía muestra los cambios miopáticos6.
La American Academy of Neurology y el Practice Issues Review Panel de la American Association of Neuromuscular and Electrodiagnostic Medicine publicaron unas pautas basadas en la evidencia para el diagnóstico y tratamiento de la distrofia muscular de cinturas y la distrofia muscular distal16. Entre las recomendaciones de estas pautas para un paciente en que se sospecha DMC se pueden citar:
-
Establecer el patrón de debilidad muscular.
-
Establecer el probable patrón hereditario.
-
Identificar las características distintivas, como la existencia de complicaciones, como la miocardiopatía.
-
Realizar pruebas genéticas específicas.
-
Si no se establece el diagnóstico, realizar una biopsia muscular.
El diagnóstico diferencial primero debe excluir las causas más frecuentes de debilidad muscular de predominio proximal. Entre ellas se encuentran las distrofias musculares de Duchenne y de Becker; causas tóxicas, endocrinológicas y autoinmunitarias de trastornos musculares; y trastornos no musculares, como miastenia grave y atrofia muscular espinal6.
La Jain Foundation ha colgado en internet (www.jain-foundation.org/alda/) un algoritmo de acceso gratuito que ayuda a definir los subtipos más frecuentes de DMC.
Posibilidades de tratamientoLa DMC es progresiva y actualmente el tratamiento es de apoyo, sin que haya tratamientos modificadores de la DMC. Tratar a estos pacientes requiere un enfoque interdisciplinario holístico con terapia (fisioterapia, terapia ocupacional, terapia respiratoria, logopedia y terapia de deglución) y atención por parte de especialistas en cardiología, neumología y traumatología cuando sea necesario. Se aconseja a los pacientes que busquen atención sanitaria en centros con experiencia en trastornos neuromusculares6.
La terapia se recomienda para mejorar la fuerza, aliviar los síntomas y signos, y promover una calidad de vida óptima. Además del ejercicio con regularidad, como levantamiento de pesas, fisioterapia y terapia ocupacional, es fundamental ayudar a los pacientes a mantener un estilo de vida lo más independiente posible13. Nadar es el ejercicio más beneficioso para los pacientes11.
La masoterapia es una posibilidad para ayudar a relajar los músculos contraídos y mejorar la flexibilidad articular. Los baños calientes son beneficiosos de una manera similar a la de los masajes. Seguir una dieta bien equilibrada y mantener un peso óptimo ayuda a evitar tensiones innecesarias en los músculos debilitados13.
Investigación en cursoLa investigación que se está llevando a cabo consiste en terapia génica, trasplante de mioblastos, anticuerpos neutralizadores de miostatina y la hormona de crecimiento e238. Solo deben ofrecerse a aquellos pacientes que se hayan inscrito en un estudio de investigación porque aún no se ha establecido su eficacia y seguridad a largo plazo16.
Las personas con DMC tienen ausencia hereditaria de la proteína distrofina. La distrofina es una proteína citoplasmática en forma de bastón, una parte vital de un complejo proteínico que conecta el citoesqueleto de una fibra muscular con la matriz extracelular adyacente a través de la membrana celular. Este complejo se conoce como el complejo proteínico asociado con la distrofina.

Las miofibrillas se destruyen y se pierde la capacidad regeneradora
Se reduce el número de fibras musculares (atrofia)
El tejido muscular se reemplaza por grasa y tejido conjuntivo, lo que inhibe las contracciones
A pesar del diagnóstico de DMC2I, Julie, de 9 años, actualmente puede estar en el colegio todo el día y sin ayuda. Aunque le cuesta correr y saltar a causa de la debilidad muscular, le gusta practicar algunos deportes, como natación, equitación y golf, a pesar de las importantes dificultades que entraña su práctica. Sin embargo, las limitaciones físicas ahora le impiden participar en otras actividades que le gustan, como el baile, el baloncesto, el tenis y el sóftbol. A Julie los ejercicios normales, como caminar largas distancias o subir escaleras, le cuestan. Cuando hace frío, Julie sufre rigidez muscular, por lo que su flexibilidad articular es limitada. Por lo general, un baño caliente con sales de Epsom, un masaje antes de ir a dormir y los estiramientos alivian las pantorrillas tensas, el dolor muscular después de realizar alguna actividad, ocasionales dolores de cadera y rodilla, la debilidad general y la dificultad de subir y bajar escaleras.
Recursos para pacientes y familias
Estos grupos prestan asistencia y apoyo a las familias afectadas por la DMC:
-
Coalition to Cure Calpain 3 (LGMD2) www.curecalpain3.org/
-
The Family Group of Beta-sarcoglycanopathy (GFB ONLUS) www.lgmd2e.org
-
Italian Association of Emery Dreifuss Muscular Dystrophy www.aidmed.org
-
Jain Foundation www.jain-foundation.org
-
Kurt + Peter Foundation www.kurtpeterfoundation.org
-
LGMD2D Foundation www.lgmd2d.org
-
The LGMD2i Research Fund www.lgmd2ifund.org
-
McColl-Lockwood Laboratory for Muscular Dystrophy Research www.carolinashealthcare.org/service.cfm?id=3193&fr=true
-
The Muscular Dystrophy Association (MDA) https://www.mda.org/
-
Nationwide Children's: Muscular Dystrophy Clinic www.nationwidechildrens.org/muscular-dystrophy-clinic
-
Patients⿿ Association for Dysferlinopathy (MMD/LGMD2B/DACM) Japan www.padj.jp/index.html
Puesto que cada paciente con DMC se ve afectado de manera diferente, los esfuerzos en la instrucción se deben adaptar para satisfacer las necesidades de cada individuo. Las enfermeras deben enseñar a los pacientes y a sus cuidadores la importancia de seguir el régimen de tratamiento prescrito y colaborar con el médico de atención primaria o la enfermera especialista. Entre los temas que deben enseñarse pueden citarse:
-
Seguir los tratamientos de fisioterapia y terapia ocupacional.
-
Mantener una dieta sana y bien equilibrada.
-
Mantener un peso saludable.
-
Realizar buenas técnicas de estiramiento.
-
Evitar caídas.
-
Garantizar el descanso y sueño necesarios.
-
Utilizar los dispositivos de asistencia, como un bastón o ⿿dispositivo⿿ de mango largo, cuando avance la debilidad.
-
Utilizar una silla de ruedas eléctrica o un scooter para ganar independencia, reducir el cansancio y evitar caídas.
-
Buscar el apoyo de un cardiólogo, neurólogo, traumatólogo y neumólogo, como se ha indicado, para que realice una evaluación de forma periódica y le ofrezca sus conocimientos.
-
Buscar apoyo emocional y orientación.
-
Realizar diferentes actividades deportivas para centrarse en la resistencia para promover la independencia en las actividades de la vida diaria10.
Las intervenciones clave de enfermería intentan proporcionar apoyo emocional y espiritual, así como instrucción a familias y pacientes. Informar de los servicios de apoyo y utilizar la escucha activa ayudará a los pacientes a enfrentarse a las dificultades y las inquietudes que genera la DMC. Las enfermeras pueden colaborar con la atención pastoral para ayudar a los pacientes y a sus familias a sobrellevar los problemas, ya que saben cómo sus vidas cambian cuando reciben este diagnóstico limitante y de por vida (v. el cuadro Recursos para pacientes y familias).
Un futuro mejorAl conocer la DMC y mantenerse al corriente de los avances en investigación, las enfermeras podrán cuidar de manera más integral a los pacientes con DMC, y mejorar la calidad de vida y los resultados de los pacientes.
Kaitlyn E. Sahd es enfermera titulada en la UCI pediátrica intermedia en el Pennsylvania State Children's Hospital en Hershey, Pennsylvania. Lisa Ruth-Sahd es profesora titular de enfermería en el York College of Pennsylvania en York, Pensilvania, y coordinadora externa de enfermería de verano en el Lancaster General Hospital en Lancaster, Pennsylvania. Keith G. Brazzo es podiatra en Foot and Ankle Associates de Lancaster, Pennsylvania.