




El síndrome de POEMS (polineuropatía, organomegalia, endocrinopatía, proteína monoclonal y alteraciones cutáneas) es un síndrome paraneoplásico infrecuente que ocurre en el contexto de una gammapatía monoclonal. Presentamos el caso de una paciente de 57 años que se presenta a nuestro servicio de neurología con una neuropatía periférica sensitivo-motora, simétrica y distal de 13 meses de evolución, en quien la constelación de hallazgos clínicos y paraclínicos nos lleva a confirmar el diagnóstico de síndrome de POEMS. Se realiza una revisión bibliográfica de los principales aspectos que deberían llevar al neurólogo a la sospecha de dicho síndrome.
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein and skin changes) is a rare paraneoplastic disorder that occurs in the setting of an underlying monoclonal gammopathy. It's characterised by multisystemic manifestations which exceed the ones originally described in the acronym. We report the case of a 57 year old female who presented at our neurology department with a 13 months history of sensory-motor, distal and symmetric peripheral neuropathy in whom clinical and ancillary evaluation confirmed the diagnosis of POEMS syndrome. We introduce a bibliographical review of the main aspects that should prompt the suspicion of this entity at the neurology consult.
El síndrome de POEMS es un síndrome paraneoplásico poco frecuente secundario a una discrasia de células plasmáticas. El acrónimo fue propuesto por Bardwick en 19801 y describe algunas de sus características: polineuropatía, organomegalia, endocrinopatía, proteína monoclonal y alteraciones cutáneas. Sin embargo, actualmente se lo reconoce como un síndrome multisistémico cuyas manifestaciones exceden al cuadro inicialmente descrito. Su incidencia y prevalencia se desconoce en poblaciones occidentales, siendo estimada en la población japonesa en 0,3 por 100.000 habitantes2.
La fisiopatología de este síndrome no está completamente aclarada, siendo reconocida una compleja interacción entre el factor de crecimiento vascular endotelial (VEGF) y diversas citoquinas proinflamatorias (IL6, TNF-alfa, IL12 e IL1) en respuesta al trastorno subyacente de las células plasmáticas3. El VEGF se encuentra elevado en pacientes con POEMS, sin embargo, sus manifestaciones no se explicarían únicamente por este hecho y su inhibición con el fármaco específico bevacizumab no ha demostrado resultados terapéuticos positivos4. Se han descrito además factores genéticos asociados a este síndrome5.
En cuanto a su presentación clínica la polineuropatía es la característica dominante y suele ser el síntoma de inicio. En la serie de 51 pacientes publicada por Nasu et al. se encontró que el 60% de los pacientes tuvo diagnóstico previo de polirradiculoneuropatía inflamatoria desmielinizante crónica (CIDP), habiendo recibido tratamiento con inmunoglobulina endovenosa6. Esta demora diagnóstica podría implicar riesgos para el paciente en cuanto a progresión de discapacidad y mortalidad, sin embargo esta asociación no ha sido formalmente estudiada.
Dada la baja prevalencia de la enfermedad, formas de presentación incompleta y su frecuente comienzo neurológico, resulta de gran importancia el reconocimiento de este tipo de casos, con el objetivo de evitar el retraso diagnóstico y, por ende, terapéutico7. Proponemos destacar aquellos aspectos que deberían llevar al neurólogo clínico a la sospecha de dicho síndrome.
Caso clínicoSe trata de una mujer de 57 años sin antecedentes de relevancia que presenta un cuadro progresivo de 4 meses de evolución de síntomas sensitivos con dolor al nivel distal de ambos miembros inferiores, agregando en la evolución compromiso motor de predominio distal, con necesidad de asistencia bilateral para la deambulación. Previo a nuestro contacto con la paciente la misma recibió evaluación en otra institución en la cual se le realiza diagnóstico de CIDP y se instaura tratamiento con inmunoglobulina intravenosa, completando 4 administraciones de 6 previstas a una dosis total de 2mg/kg. El estudio de neuroconducción en dicho momento informa una neuropatía periférica crónica con compromiso mielínico y axonal. Si bien la paciente refiere mejoría del dolor bajo este tratamiento, agrega compromiso motor y sensitivo de ambos miembros superiores durante el mismo. Concomitantemente presenta adelgazamiento de 30kg.
La paciente llega a nuestro servicio a los 13 meses de iniciado el cuadro; en la primera evaluación al examen neurológico destacamos la presencia de hipotonía de 4 miembros, atrofia muscular en las manos, el cuádriceps, tibial anterior y en los gastrocnemios con relativa simetricidad. Las fuerzas musculares se encontraban globalmente afectadas, siendo el compromiso severo en los extensores del carpo y de los dedos y en los flexores de los dedos. En los miembros inferiores destacamos la plejía en la dorsiflexión del pie bilateral con leve afectación proximal. Se resumen en la tabla 1 los hallazgos en cuanto al compromiso motor. Asocia arreflexia global y déficit sensitivo para todas las modalidades en las 4 extremidades, simétrico y de severa entidad en los miembros inferiores. Destacamos el hallazgo de papiledema bilateral con bordes sobreelevados y angioesclerosis. No presenta compromiso craneal ni dolor en dicho momento.
Hallazgos respecto al compromiso motor
| Al inicio del tratamiento | 7 meses bajo tratamiento | |||
|---|---|---|---|---|
| Músculo | Derecha | Izquierda | Derecha | Izquierda |
| Flexores de cuello | 5 | 5 | 5 | 5 |
| Trapecio | 5 | 5 | 5 | 5 |
| Deltoides | 5 | 5 | 5 | 5 |
| Bíceps | 4 | 4 | 5 | 4 |
| Tríceps | 4 | 4+ | 5 | 5 |
| Extensores del carpo | 1 | 1 | 4 | 4- |
| Extensores de los dedos | 1 | 1 | 2 | 2 |
| Flexores de los dedos | 4 | 4 | 4 | 4 |
| Aductor del pulgar | 2 | 2 | 2 | 2 |
| Interóseos dorsales | 0 | 0 | 0 | 0 |
| Psoas | 5 | 4+ | 5 | 5 |
| Cuádriceps | 4 | 4- | 5 | 5 |
| Bíceps crural | 4+ | 4 | 5 | 5 |
| Aductores | 5 | 5 | 5 | 5 |
| Gastrocnemios | 4- | 0 | 5 | 5 |
| Tibial anterior | 0 | 0 | 1 | 1 |
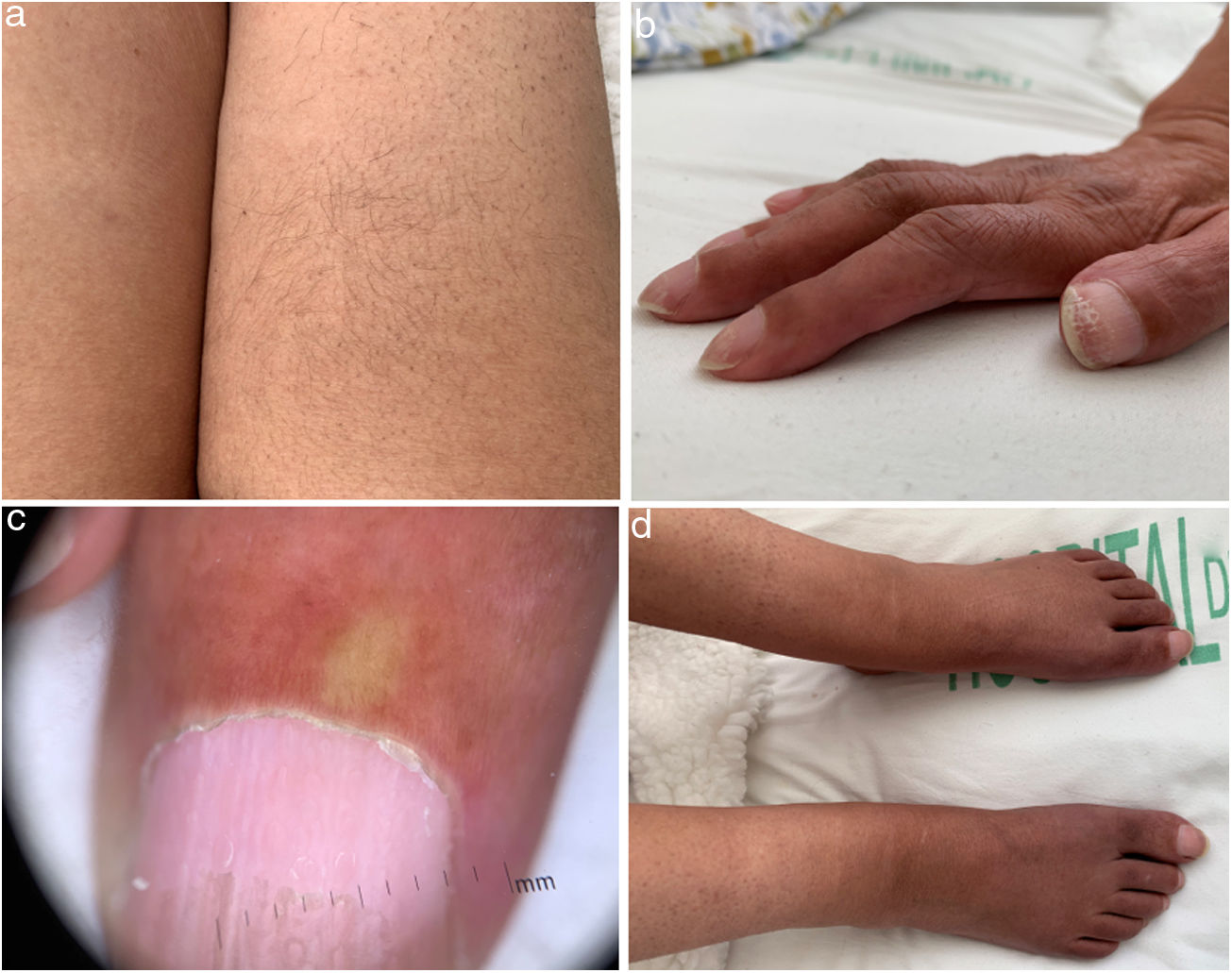
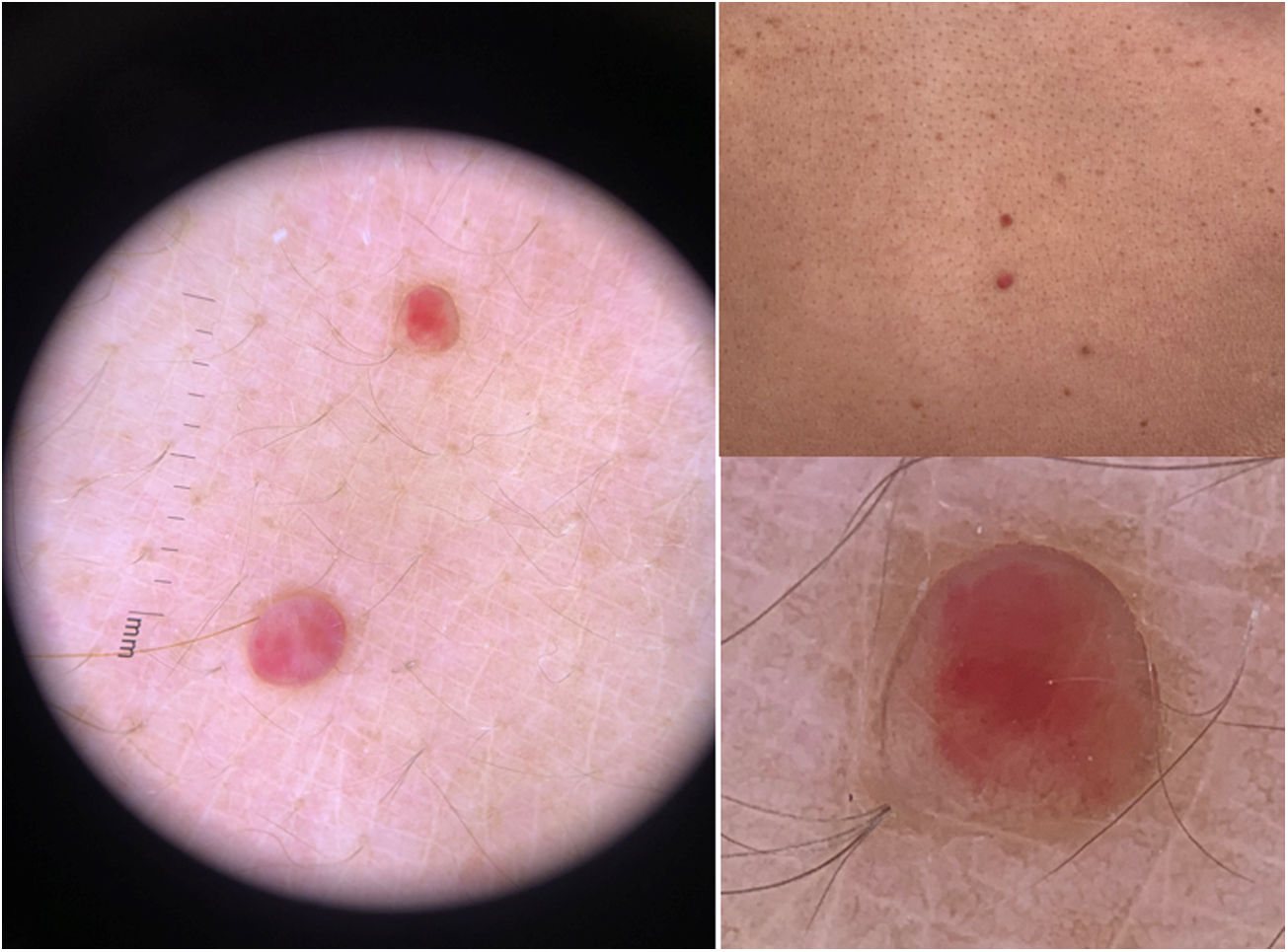
Del examen extraneurológico la paciente se presenta adelgazada, con un aceptable estado general. El examen clínico cutáneo reveló la presencia de acropaquia en los dedos de las manos y los pies, cambios esclerodermiformes en los dedos, como la presencia de piel esclerótica, la pérdida de vellos y la alteración en la dermatoscopia del lecho ungueal (presencia de espacios avasculares y disminución del número de capilares). En los miembros inferiores, al nivel de las piernas, presentaba edema blando, y al nivel de la cara se destaca una lipoatrofia facial (la paciente relata la aparición junto con el cuadro neurológico) (fig. 1). También se destaca la presencia de hiperpigmentación difusa, hipertricosis y en la cara anterior del tórax presentaba 2 lesiones papulares rojas que se correspondían con hemangiomas glomerulares (fig. 2) confirmados por histopatología.
 Presencia de hipertricosis. b) Acropaquia, acrocianosis y cambios esclerodermiformes en manos. c) Alteración en la dermatoscopia del lecho ungueal (presencia de espacios avasculares y disminución del número de capilares). d) Edema blando en MMII e hiperpigmentación.")

Se realiza un nuevo estudio de neuroconducción donde se observa una polineuropatía sensitivo-motora severa con compromiso de los 4 miembros, siendo la mayoría inexcitables. Al nivel del nervio axilar se evidencia compromiso mielínico y en los miembros inferiores denervación axonal activa. Dado el severo compromiso, tanto clínico como electrofisiológico, y la ausencia de respuesta al tratamiento previamente instaurado se decide realizar una nueva búsqueda etiológica, de la cual se destaca el hallazgo de una hipoproteinemia global con una banda de aspecto monoclonal de 0,5g/dl en la zona de migración de las betaglobulinas, con una inmunofijación positiva IgA lambda, planteándose el diagnóstico de síndrome de POEMS. En el rodado de médula ósea se observan plasmocitos del 7%, algunas formas binucleadas y plasmoblastos. En el inmunofenotipo presenta 0,34% plasmocitos, 81% de los cuales eran aberrantes y clonales. En la biopsia de médula ósea se destaca una población de células de morfología plasmocitaria que representan un 15% de la celularidad, de topografía perivascular e intersticial, individuales, aisladas y formando pequeños agregados con perfil IHQ CD138+, CD20–, CD3– y CD52– y franco predominio de expresión de cadenas livianas lambda, compatible con un SMM (mieloma quiescente). Se solicita determinación de VEGF siendo superior a 700pg/ml. Dosificación de Ig: IgG 677mg/dl, IgA e IgM en rango. Cadenas livianas libres en suero normales.
Destacamos además la presencia de hepatoesplenomegalia y ascitis en la tomografía, y la ausencia de lesiones osteoescleróticas. Se resumen en la tabla 2 los hallazgos paraclínicos complementarios. Se inició tratamiento con melfalán 9mg/m2 día por 4 días asociado a dexametasona 40mg vía oral por 4 días cada 6 semanas. En la última consulta de seguimiento, a los 7 meses de iniciado el tratamiento, se evidencia una mejoría en la fuerza muscular de varios de los músculos evaluados (tabla 1), notándose además una mayor autonomía funcional referida por la paciente. No contamos con estudio neurofisiológico luego del tratamiento. Desde el punto de vista hematológico presentó un descenso en los niveles de VEGF, siendo de 44pg/ml en el último control, demostrando así una respuesta al tratamiento instaurado.
Hallazgos paraclínicos complementarios
| Hematológicos | Hemoglobina | 16,8g/dl |
| Hematocrito | 52,9% | |
| Plaquetas | 298.000/mm3 | |
| Leucocitos | 10.540/mm3 | |
| JAK2 | Negativo | |
| Endocrinológicos | Glucemia | 83mg/dl |
| HbA1c | 4,9% | |
| Vitamina B12 | 182pg/ml | |
| Homocisteinemia | 37mmol/l | |
| Ácido fólico | 2,9ng/ml | |
| TSH | 7,95 | |
| Anticuerpos antitiroglobulina | Negativos | |
| Estudio de LCR | Glucorraquia | 0,57g/dl |
| Proteinorraquia | 0,74g/dl | |
| Glóbulos blancos | 2 | |
| Otros | Albúmina | 4,0g/dl |
El síndrome de POEMS es un síndrome paraneoplásico infrecuente secundario a una discrasia de células plasmáticas. De acuerdo a los criterios diagnósticos propuestos por Dispenzieri es obligatoria la presencia de polirradiculoneuropatía y proliferación monoclonal de células plasmáticas; uno de los criterios mayores, como son las lesiones osteoescleróticas, VEGF elevado y/o la presencia de enfermedad de Castleman y al menos uno de los criterios menores como organomegalia, endocrinopatía, cambios en la piel, edema de papila, sobrecarga de volumen extravascular y trombocitosis o policitemia8. La paciente que hemos presentado presenta ambos criterios obligatorios, VEGF elevado como criterio mayor y 4 criterios menores (organomegalia, papiledema, alteraciones cutáneas y elementos de sobrecarga de volumen extravascular como ascitis), por lo cual confirmamos el diagnóstico de síndrome de POEMS.
Su diagnóstico temprano es difícil debido a su baja prevalencia y a la heterogeneidad de sus síntomas. Habitualmente se retrasa el diagnóstico, lo que se cree conlleva a un peor pronóstico de los síntomas neurológicos9. Se ha visto un retraso de más de 13 a 18 meses10 en los pacientes con POEMS, similar al observado en nuestra paciente. Esto se debe a las dificultades en distinguir el síndrome de POEMS del inicio de una CIDP, como también sucedió con nuestra paciente. Existen algunas claves a la hora de diferenciarlas, de utilidad para el neurólogo clínico, que veremos a continuación.
La neuropatía periférica es la carta de presentación de estos pacientes; es habitualmente de inicio subagudo, compromiso sensitivo-motor, simétrica, de inicio distal y de progresión ascendente, al igual que en nuestra paciente. Los miembros inferiores se afectan antes y de forma más severa que los miembros superiores, y los síntomas sensitivos generalmente preceden a los motores. Con la progresión de la enfermedad se consolida la paresia severa distal en los miembros inferiores y superiores pudiendo llegar a ser invalidante11. El dolor neuropático es más común en pacientes con POEMS que en el caso de CIDP, por lo cual se debe considerar como un elemento de sospecha. Este estuvo presente en nuestra paciente al inicio del cuadro. Diferentes series han reportado la presencia de dolor neuropático en el 76% de los pacientes con síndrome de POEMS, en contraposición al 7% de los pacientes con CIDP6.
La neurofisiología también resulta de ayuda; en el caso del síndrome de POEMS encontramos una neuropatía sensitivo-motora primariamente desmielinizante con degeneración axonal secundaria. La afectación es largo dependiente y se caracteriza por compromiso temprano de potenciales motores con un patrón de daño predominantemente axonal en los miembros inferiores y desmielinizante en los miembros superiores. Este hallazgo, reportado por Liu et al.12, es consistente con el encontrado en nuestra paciente. Un estudio6 que comparó los perfiles neurofisiológicos entre pacientes con POEMS y CIDP concluyó que los pacientes con POEMS se diferencian de CIDP en que muestran un mayor grado de pérdida axonal, el enlentecimiento de conducción de los potenciales motores es mayor en los segmentos intermedios del nervio y raramente se encuentran bloqueos de conducción. Este estudio reportó además que el 70% de los pacientes con POEMS cumple criterios electrofisiológicos para CIDP, otro factor que debe llevar al neurólogo clínico a tener un elevado índice de sospecha a la hora de diferenciar a estos pacientes.
El examen dermatológico también resulta de gran utilidad, ya que los trastornos cutáneos están presentes en el 68% de los pacientes, siendo la hiperpigmentación cutánea difusa, la plétora y la acrocianosis las más comunes. Otras manifestaciones incluyen hiperhidrosis, leuconiquia, vasculitis necrosante, hipertricosis y calcifilaxis. También se informa engrosamiento cutáneo de tipo esclerodermiforme. Estos hechos pueden estar relacionados con una mayor deposición de colágeno por los fibroblastos dérmicos, causada por el aumento de los niveles de VEGF. La hiperpigmentación puede ser difusa o localizada, y ocurre principalmente en las superficies extensoras, el dorso, el cuello y las axilas. Suele retroceder en respuesta al tratamiento. La leuconiquia es una manifestación bastante rara13.
La aparición de múltiples angiomas cutáneos en el síndrome de POEMS se considera infrecuente. No obstante, se ha documentado en el 25% al 45% de los pacientes, apareciendo durante el curso de la enfermedad como lesiones papulares firmes, con coloración eritematosa o violeta, en el tronco y en la región proximal de las extremidades. Por lo general, presentan características histológicas variables, siendo el hemangioma capilar lobular más frecuente y con menor frecuencia el hemangioma glomerular. La histopatología del hemangioma glomerular muestra un enredo capilar que ocupa un gran espacio vascular en la dermis llena de células endoteliales, con un aspecto que se asemeja a un glomérulo renal. Está presente en solo el 3% de los casos y puede preceder a los signos y síntomas restantes del síndrome, lo que permite un diagnóstico precoz14.
Otro elemento de utilidad es la presencia de papiledema, como se vio en esta paciente. Este suele ser bilateral y es un signo temprano en el curso de la enfermedad. Una serie15 de 94 pacientes con síndrome de POEMS encontró una prevalencia del 52,1%, estando asociado a niveles elevados de proteínas en el líquido cefalorraquídeo. En nuestra paciente el estudio del LCR evidenció la presencia de disociación albúmino-citológica, que se ha reportado en pacientes con POEMS, siendo no específica, ya que la encontramos en otras entidades como la CIDP16.
Si bien en esta paciente los niveles tanto de ácido fólico como de vitamina B12 se encontraban por debajo del rango, tanto las características clínicas como neurofisiológicas no son concordantes con un cuadro neurológico causado por dichos déficits. Además existen múltiples reportes de déficit de vitamina B12 en pacientes con síndrome de POEMS17.
La ausencia de respuesta al tratamiento estándar de la CIDP también nos debe llevar a ahondar en la búsqueda de otras etiologías, teniendo en cuenta que la polineuropatía es la complicación más común de las gammapatías monoclonales y que en un 10% de los pacientes que se presentan con polineuropatía se encontrará una discrasia de células plasmáticas18, por lo cual su búsqueda no debe omitirse en este tipo de pacientes19. La presencia de gammapatía monoclonal IgA o IgG tipo λ es esencial para el diagnóstico de POEMS. Existen varias entidades que pueden causar neuropatía y asociar un pico monoclonal, con las cuales se debe realizar diagnóstico diferencial (gammapatía monoclonal de significado incierto, mieloma múltiple, macroglobulinemia de Waldenström, amiloidosis primaria, etc.) dadas las implicanciones terapéuticas de dichos diagnósticos20. En el caso de nuestra paciente se confirma por biopsia de médula ósea la presencia del 15% de células plasmáticas clonales, clasificando la enfermedad de base dentro de las discrasias plasmocitarias como un SMM, ya que cumple con el criterio de porcentaje de plasmocitos mayor al 10% en la médula ósea, clonales por inmunofenotipo e inmunohistoquímica, sin hechos CRAB (hipercalcemia, fallo renal, anemia y lesiones óseas líticas). Cabe destacar que tampoco cumple con otros criterios definitorios de mieloma múltiple, que son el porcentaje de infiltración medular mayor al 60%, 2 o más lesiones líticas óseas mayores a 5mm confirmadas por resonancia magnética o ratio de FLC mayor a 100.
En cuanto al tratamiento, el mismo se basa en disminuir el porcentaje de células plasmáticas causantes del aumento del VEGF. Este tratamiento demostró ser más efectivo que bajar los niveles de VEGF directamente, ya que ensayos clínicos con bevacizumab, un anticuerpo monoclonal dirigido al VEGF no han arrojado resultados consistentemente positivos8. No existen estudios aleatorizados que comparen planes terapéuticos, por lo que las recomendaciones se hacen de casos reportados o experiencia en algunos centros. El tratamiento dependerá de la localización de la enfermedad. Cuando el compromiso es sistémico el uso de melfalán y dexametasona logra un 81% de respuestas globales con un 100% de descenso del VEGF y respuesta neurológica variable. El uso de quimioterapia a altas dosis, asociado a trasplante autólogo de progenitores hematopoyéticos, ha mostrado que el 100% de los pacientes refirió mejoría neurológica. La sobrevida global a 10 años es del 90%. Sin embargo, este tratamiento se ha asociado a mayor toxicidad. El uso de lenalidomida con dexametasona por 12 ciclos logró el 95% de respuestas neurológicas con sobrevida global a los 3 años del 90%21.
En cuanto al pronóstico, un estudio retrospectivo realizado por la Clínica Mayo22 encontró que la menor edad, niveles de albúmina mayores a 3,2g/dl y la presencia de respuesta hematológica completa al tratamiento de primera línea se asocian a mayor sobrevida. Las tasas de sobrevida varían de acuerdo a la modalidad terapéutica seleccionada. En un estudio realizado en 361 pacientes en China la tasa de sobrevida del global a los 5 y 10 años fue del 84% y del 77% respectivamente, reportando este grupo la asociación con menor sobrevida y la presencia de edad>50 años, hipertensión pulmonar, derrame pleural y/o <30ml/min/1,73m2 de filtrado glomerular23. Teniendo en cuenta la globalidad de los factores de mal pronóstico, en nuestra paciente encontramos únicamente la edad. En cuanto al pronóstico funcional un estudio24 realizado por Misawa et al. encontró que la normalización del VEGF a los 6 meses es un predictor para el tiempo de sobrevida sin actividad, así como para la mejoría de la función motora de los MMSS y de los marcadores neurofisiológicos. En la evolución en los primeros 6 meses de tratamiento con melfalán y dexametasona cada 6 semanas a dosis ya mencionadas, nuestra paciente mostró una notable disminución del VEGF (demostrando así de manera indirecta la disminución de infiltración por células plasmáticas) y por consiguiente mejoría en lo neurológico, desaparición de las manifestaciones cutáneas y ascitis, persistiendo aún las organomegalias. Cabe destacar además la mejoría en la calidad de vida y salud psicosocial de la paciente, sabiendo que se trata de una paciente joven, activa hasta el diagnóstico.
La heterogeneidad clínica e infrecuencia del síndrome de POEMS son aspectos que llevan a que su diagnóstico sea un verdadero reto. Se presentó el caso de una paciente que comienza con una polineuropatía con elementos atípicos para el planteamiento de CIDP y múltiples indicadores de afectación sistémica, en la cual se confirmó el diagnóstico de síndrome de POEMS. Algunos aspectos como la presencia de dolor, el compromiso mielínico con lesión axonal marcada en los miembros inferiores, el patrón largo-dependiente y la presencia de papiledema, alteraciones cutáneas y organomegalia, entre otros, son algunas de las pistas que nos deben hacer sospechar la presencia de una gammapatía monoclonal como causante del cuadro, destacando las implicaciones terapéuticas y pronósticas de la amplia constelación de diferenciales. El abordaje interdisciplinario es fundamental en estos casos (neurólogos, hematólogos, fisiatras, psicólogos y asistentes sociales) para lograr un diagnóstico precoz, un inicio de tratamiento adecuado de forma temprana y manejo de complicaciones en vistas a obtener una mejoría de la calidad de vida y pronóstico, tanto vital como funcional.
Conflicto de interesesSe deja constancia de que no tenemos conflicto de intereses.
