



Guillain-Barré syndrome (GBS) is an acute-onset, immune-mediated disease of the peripheral nervous system. It may be classified into 2 main subtypes: demyelinating (AIDP) and axonal (AMAN). This study aims to analyse the mechanisms of axonal damage in the early stages of GBS (within 10 days of onset).
DevelopmentWe analysed histological, electrophysiological, and imaging findings from patients with AIDP and AMAN, and compared them to those of an animal model of myelin P2 protein–induced experimental allergic neuritis. Inflammatory oedema of the spinal nerve roots and spinal nerves is the initial lesion in GBS. The spinal nerves of patients with fatal AIDP may show ischaemic lesions in the endoneurium, which suggests that endoneurial inflammation may increase endoneurial fluid pressure, reducing transperineurial blood flow, potentially leading to conduction failure and eventually to axonal degeneration. In patients with AMAN associated with anti-ganglioside antibodies, nerve conduction block secondary to nodal sodium channel dysfunction may affect the proximal, intermediate, and distal nerve trunks. In addition to the mechanisms involved in AIDP, active axonal degeneration in AMAN may be associated with nodal axolemma disruption caused by anti-ganglioside antibodies.
ConclusionInflammatory oedema of the proximal nerve trunks can be observed in early stages of GBS, and it may cause nerve conduction failure and active axonal degeneration.
El síndrome de Guillain-Barré (SGB) es un trastorno agudo e inmuno-mediado del sistema nervioso periférico. Sus dos subtipos básicos son el desmielinizante (AIDP) y el axonal (AMAN). El objetivo de este trabajo es abordar los mecanismos de daño axonal en la fase precoz del síndrome (≤10 días del inicio sintomático).
DesarrolloSe han revisado aspectos histológicos, neurofisiológicos y de imagen descritos tanto en la AIDP como en la AMAN. Los hallazgos en la patología humana han sido contrastados con lo reportado en la neuritis alérgica experimental inducida por el componente P2 de la mielina. El edema inflamatorio de las raíces raquídeas y de los nervios espinales constituye la lesión inicial en el SGB. En los nervios espinales de casos fatales de AIDP se ha demostrado la presencia de lesiones isquémicas endoneurales, lo cual sugiere que la inflamación puede condicionar un incremento de su presión con reducción del flujo sanguíneo transperineural, que puede desencadenar fallo de la conducción y eventualmente degeneración axonal secundaria. En la AMAN con anticuerpos antigangliósido el bloqueo de la conducción por disfunción de los canales del sodio nodales puede afectar a troncos nerviosos proximales, intermedios y distales. Además de los mecanismos que operan en la AIDP, la degeneración axonal activa en la AMAN puede ir ligada a la disrupción del axolema nodal inducida por los anticuerpos anti-gangliósido.
ConclusiónEn la fase precoz del SGB hay edema inflamatorio de los troncos nerviosos proximales, que puede condicionar fallo de la conducción nerviosa y degeneración axonal activa.
The Barraquer Lecture at the 54th Annual Meeting of the Spanish Society of Neurology (Barcelona, 2002) addressed the complex pathophysiology of axonal degeneration in Guillain-Barré syndrome (GBS).1 This review considers the advances made in our understanding of the disease in the last 15 years, focusing on the early stages of progression, conventionally agreed to be the first 10 days after symptom onset.2
GBS can manifest with 3 basic patterns: 1) the classic pattern, acute inflammatory demyelinating polyneuropathy (AIDP); 2) acute motor axonal neuropathy (AMAN) or acute motor and sensory axonal neuropathy (AMSAN); and 3) Miller Fisher syndrome (MFS).3–7 Fundamentally, the clinical manifestations of AIDP/AMAN-AMSAN include acute, ascending, flaccid, areflexic paralysis, with severity peaking within the first 4 weeks of progression.
The annual incidence rate is 1-2 cases/100000 population, with axonal forms being the most prevalent in certain Asian countries and AIDP being the most common variant in Europe and the United States.8–10
Early Guillain-Barré syndrome: invaluable historical referencesHaymaker and Kernohan11 performed the first autopsy studies of early GBS, examining 50 patients, of whom 32 had died between days 2 and 10 after onset. The only lesion observed in the first 4 days of progression was endoneurial oedema, predominantly involving the convergence of the dorsal and ventral spinal roots, where they form the spinal nerve.
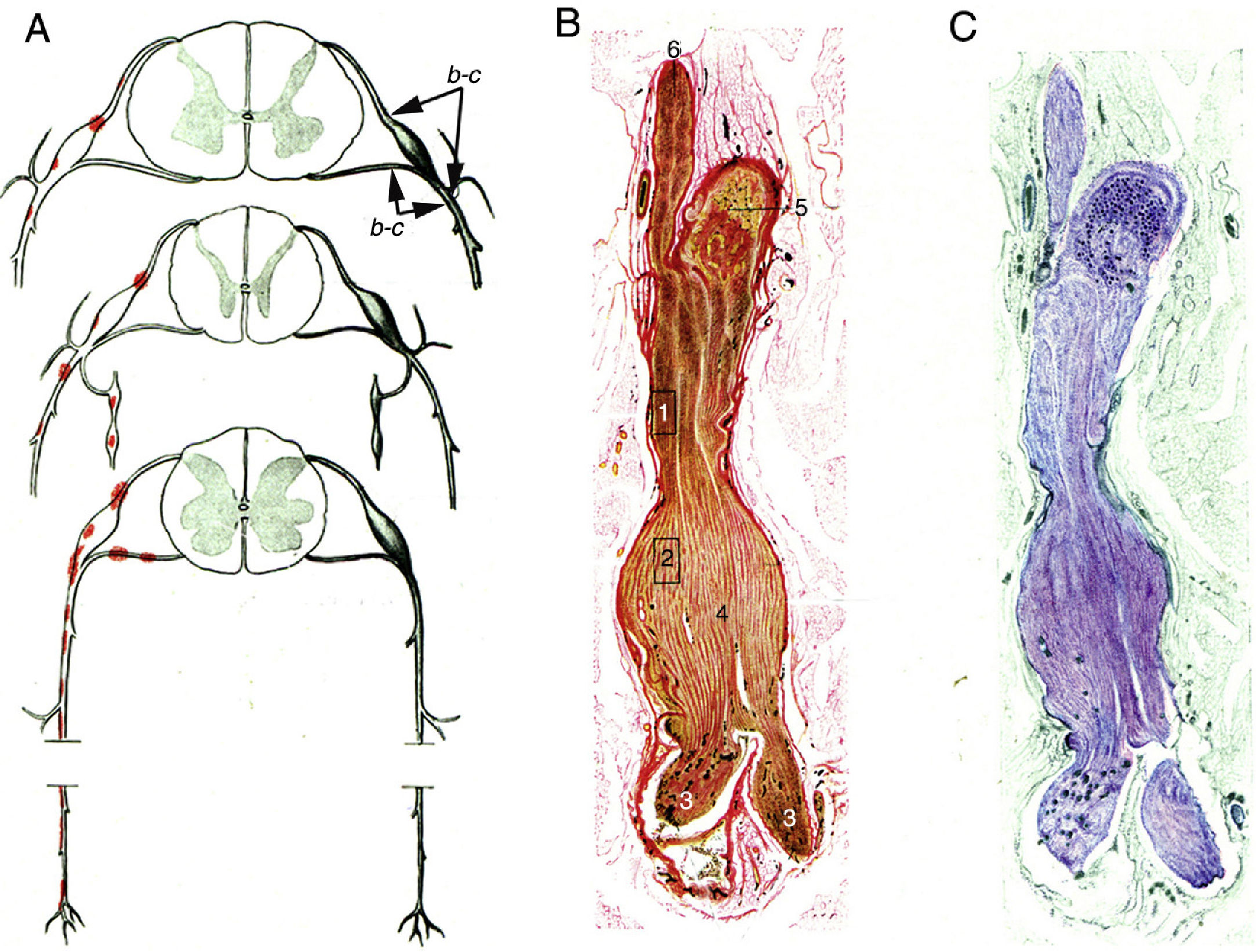
Krücke12 noted the inflammatory nature of endoneurial oedema, which appears from 24 hours after symptom onset and develops fully by day 3; this study also confirmed the predominance of inflammatory oedema of the proximal nerve trunks, particularly in the spinal nerves (Fig. 1).
 Diagram of lesion topography in GBS (from top to bottom: cervical, thoracic, and lumbar regions). Lesions (red dots) are observed in proximal nerve trunks, including the ventral and dorsal spinal roots, spinal ganglia, sympathetic ganglia, and the ventral rami of the spinal nerves. The labels b and c used by the author to signal the localisation of figures 65, 66 (here Fig. 1B and C), and 67 (not reproduced) are maintained. B) Longitudinal section of a nerve segment between the ventral spinal root and the spinal nerve, taken from a patient with GBS who died on the 18th day of progression. The original numbering is maintained. 1 and 2: areas illustrated by Krücke in other figures, demonstrating extensive “mucoid endoneurial oedema” (inflammatory); 3: spinal nerve rami (the ventral and the dorsal ramus); 4: fusiform dilation of the spinal nerve; 5: spinal ganglion; and 6: ventral spinal root. Van Gieson stain, magnification not specified. C) Another longitudinal section from the same location, showing the purple colouration of the fusiform dilation of the spinal nerve. Cresyl violet stain.")
Minimally altered reproduction of figures 65 to 67 from Krücke's12 article.
A) Diagram of lesion topography in GBS (from top to bottom: cervical, thoracic, and lumbar regions). Lesions (red dots) are observed in proximal nerve trunks, including the ventral and dorsal spinal roots, spinal ganglia, sympathetic ganglia, and the ventral rami of the spinal nerves. The labels b and c used by the author to signal the localisation of figures 65, 66 (here Fig. 1B and C), and 67 (not reproduced) are maintained.
B) Longitudinal section of a nerve segment between the ventral spinal root and the spinal nerve, taken from a patient with GBS who died on the 18th day of progression. The original numbering is maintained. 1 and 2: areas illustrated by Krücke in other figures, demonstrating extensive “mucoid endoneurial oedema” (inflammatory); 3: spinal nerve rami (the ventral and the dorsal ramus); 4: fusiform dilation of the spinal nerve; 5: spinal ganglion; and 6: ventral spinal root. Van Gieson stain, magnification not specified.
C) Another longitudinal section from the same location, showing the purple colouration of the fusiform dilation of the spinal nerve. Cresyl violet stain.
Asbury et al.13 refuted the pathogenic role of endoneurial oedema in early GBS, in a famous article that includes a clinico-pathological study of 19 patients with fatal GBS, of whom 5 died in the first 8 days of progression. According to these authors, the histological substrate of the syndrome is a perivenular mononuclear inflammatory infiltrate, of patchy distribution, with segmentary demyelination; however, they do not recognise a “state of oedema” at the macroscopic or microscopic level. The pathophysiological relevance of oedema in inflammatory neuropathies has been reconsidered in recent articles.14
We described a fulminant case of AIDP in which the patient died on the ninth day of progression.15Figs. 2 and 3 illustrate the peripheral nervous system (PNS) pathology, comprising marked inflammatory endoneurial and epi-perineurial oedema with incipient demyelination, predominantly affecting the ventral rami of the cervical and lumbar nerves.15–17
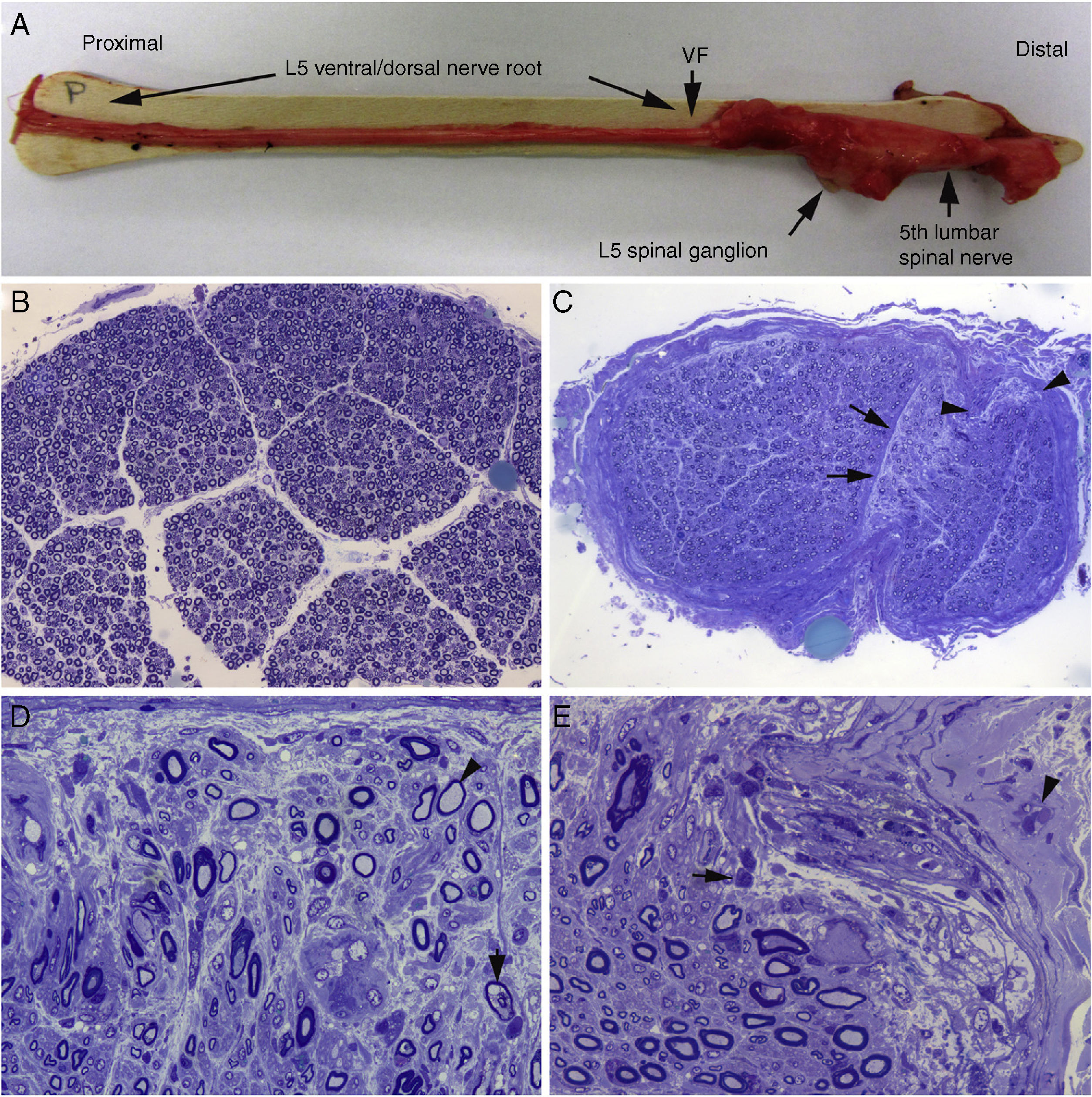
 Macroscopic view of the L5 nerve from the proximal end to its entry into the vertebral foramen (VF). The dissection continues to show the spinal ganglion and the fifth lumbar nerve. The nerve is thickened after passing through the VF (see Fig. 1B and C). B) In this semithin section of the L5 ventral root, taken 1cm beyond its passage through the VF, myelin fibre density is preserved. Toluidine blue stain; ×100 before reduction. C) In this semithin section of the ventral ramus of the fifth lumbar nerve, at the level of its emergence from the VF, we observe diffuse endoneurial oedema, which is more marked in certain subperineurial areas (arrowheads) and in one of the areas adjacent to the perineurial septum (arrows). Endoneurial oedema spaces out the myelinated fibres, hence their apparent reduced density as compared to the previous images. Toluidine blue stain; ×65 before reduction. D) Detailed image of the area beside the septum, signalled by the arrows in Fig. 2C. Inflammatory oedema is apparent, with numerous mononuclear cells, isolated fibres with inappropriately thin myelin sheaths (arrowhead), and fibres presenting vacuolar myelin degeneration (arrow). Toluidine blue stain; ×630 before reduction. E) Detailed image of the subperineurial area signalled with arrowheads in Fig. 2C. Marked oedema is observed, with endoneurial and epineurial inflammatory cells, (arrow and arrowhead, respectively). Toluidine blue; ×630 before reduction.")
Histological findings in a fatal case of AIDP.15
A) Macroscopic view of the L5 nerve from the proximal end to its entry into the vertebral foramen (VF). The dissection continues to show the spinal ganglion and the fifth lumbar nerve. The nerve is thickened after passing through the VF (see Fig. 1B and C).
B) In this semithin section of the L5 ventral root, taken 1cm beyond its passage through the VF, myelin fibre density is preserved. Toluidine blue stain; ×100 before reduction.
C) In this semithin section of the ventral ramus of the fifth lumbar nerve, at the level of its emergence from the VF, we observe diffuse endoneurial oedema, which is more marked in certain subperineurial areas (arrowheads) and in one of the areas adjacent to the perineurial septum (arrows). Endoneurial oedema spaces out the myelinated fibres, hence their apparent reduced density as compared to the previous images. Toluidine blue stain; ×65 before reduction.
D) Detailed image of the area beside the septum, signalled by the arrows in Fig. 2C. Inflammatory oedema is apparent, with numerous mononuclear cells, isolated fibres with inappropriately thin myelin sheaths (arrowhead), and fibres presenting vacuolar myelin degeneration (arrow). Toluidine blue stain; ×630 before reduction.
E) Detailed image of the subperineurial area signalled with arrowheads in Fig. 2C. Marked oedema is observed, with endoneurial and epineurial inflammatory cells, (arrow and arrowhead, respectively). Toluidine blue; ×630 before reduction.
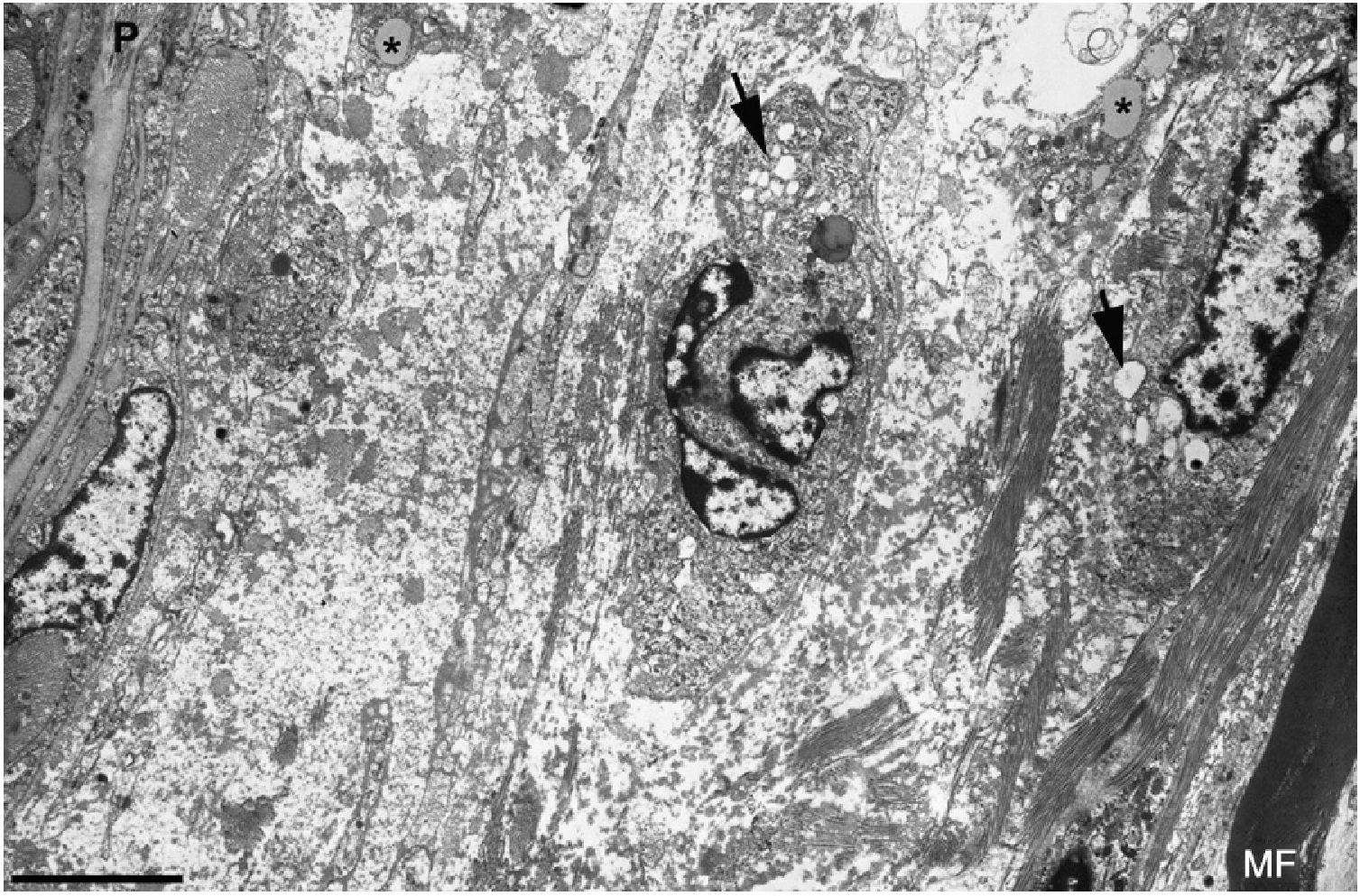
 and numerous endocytic vesicles (arrows) and lysosomes. The oedematous area does not contain myelinated fibres; the only one observed (MF) is situated 20μm from the perineurium (P). Scale bar: 3μm.")
Ultrastructural image of the subperineurial region shown in Fig. 2E. We observe marked endoneurial oedema with amorphous material, probably proteoglycans, and disperse collagen fibril bundles. Note the presence of macrophages containing lipid droplets (asterisks) and numerous endocytic vesicles (arrows) and lysosomes. The oedematous area does not contain myelinated fibres; the only one observed (MF) is situated 20μm from the perineurium (P). Scale bar: 3μm.
In summary, inflammatory oedema of the proximal nerve trunks is of great importance in early GBS.
Experimental allergic neuritis: key pointsExperimental allergic neuritis (EAN), which can be induced by various antigens (eg, P0, P2, PMP22, or glycolipids), is used as an animal model of GBS.18,19 In this section, we address certain histological and neurophysiological aspects of the initial phase of EAN induced by P2 myelin protein (P2-EAN).
In Lewis rats, administration of 50μg of SP26 (residues 53-76 of bovine P2 myelin protein) induces pure inflammatory demyelination.20 If this dose is doubled, the inflammatory demyelination of the nerve roots remains unchanged, but is replaced by axonal degeneration in the sciatic nerve, with mainly centrofascicular involvement.21 This change in lesion topography is related to the ubiquitous endoneurial inflammation by a bystander effect. Additionally, the centrofascicular Wallerian degeneration observed suggests the involvement of ischaemic events.22 In a sequential teased fibre study of another EAN model, only a small percentage of fibres displaying inflammatory demyelination (5%-10%) presented axonal degeneration23; this brings the pathogenic role of the bystander effect into question.
In P2-EAN induced by transfer of CD4+ lymphocytes, the only lesion affecting the nerve trunks at symptom onset (post-inoculation day 4) is endoneurial and epineurial inflammatory oedema.24 When conventional doses of CD4+ cells are transferred, electrophysiological findings on the fifth day indicate demyelination.25 If the dose is increased, the electrophysiological pattern progresses to complete nerve conduction failure, which the authors attribute to Wallerian degeneration. This interpretation is certainly debatable, as motor nerve excitability is only lost 8-9 days after nerve section in Wallerian degeneration.26,27
Histological studies of P2-EAN rats have shown that axonal degeneration appears at the peak of development of endoneurial inflammatory oedema and the increase in endoneurial pressure.28
In summary, P2-EAN may involve a dissociated process comprising radicular demyelination and active axonal degeneration in more distal nerve trunks.
Topographical variations in the blood-nerve barrierThe blood-nerve barrier plays an essential role in the homeostasis of the PNS.29,30 The endothelium of endoneurial capillaries has occluding junctions and is surrounded by a basal membrane and pericytes. Experimental studies have demonstrated topographical differences in PNS vascular permeability, with greater permeability in the spinal ganglia and the ventral and dorsal spinal nerve roots until the junction with the spinal nerve.31 Therefore, the blood-nerve barrier at nerve terminals is known to be less efficient.32
These characteristics of the blood-nerve barrier explain the selective vulnerability of the spinal roots, spinal ganglia, spinal nerves, and nerve terminals to autoimmune attacks, particularly those mediated by antibodies.19,33
Axonal Guillain-Barré syndromeGBS has classically been considered a prototypical example of acute inflammatory demyelinating neuropathy; in other words, GBS and AIDP were used interchangeably in reference to a single syndrome with varying degrees of secondary axonopathy.13,34–36
In 1986, Feasby et al.37 presented 5 patients with severe GBS, with initial neurophysiological findings showing inexcitability of motor and sensory nerves. In 3 cases (patients 1, 3, and 5), inexcitability was detected between days 2 and 5 of progression. The autopsy of patient 1 revealed primary axonal degeneration without inflammation or demyelination; this led to the recognition of axonal GBS. It may be argued that purely axonal involvement would not explain such early loss of nerve excitability.
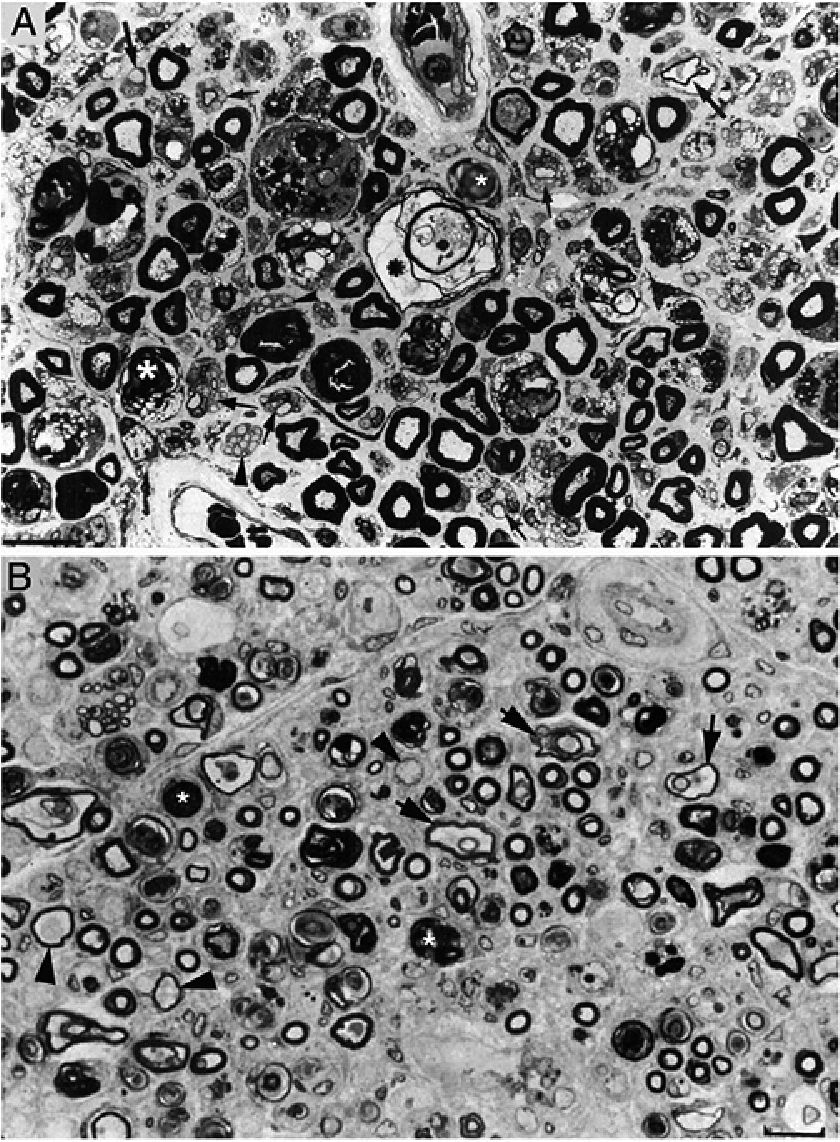
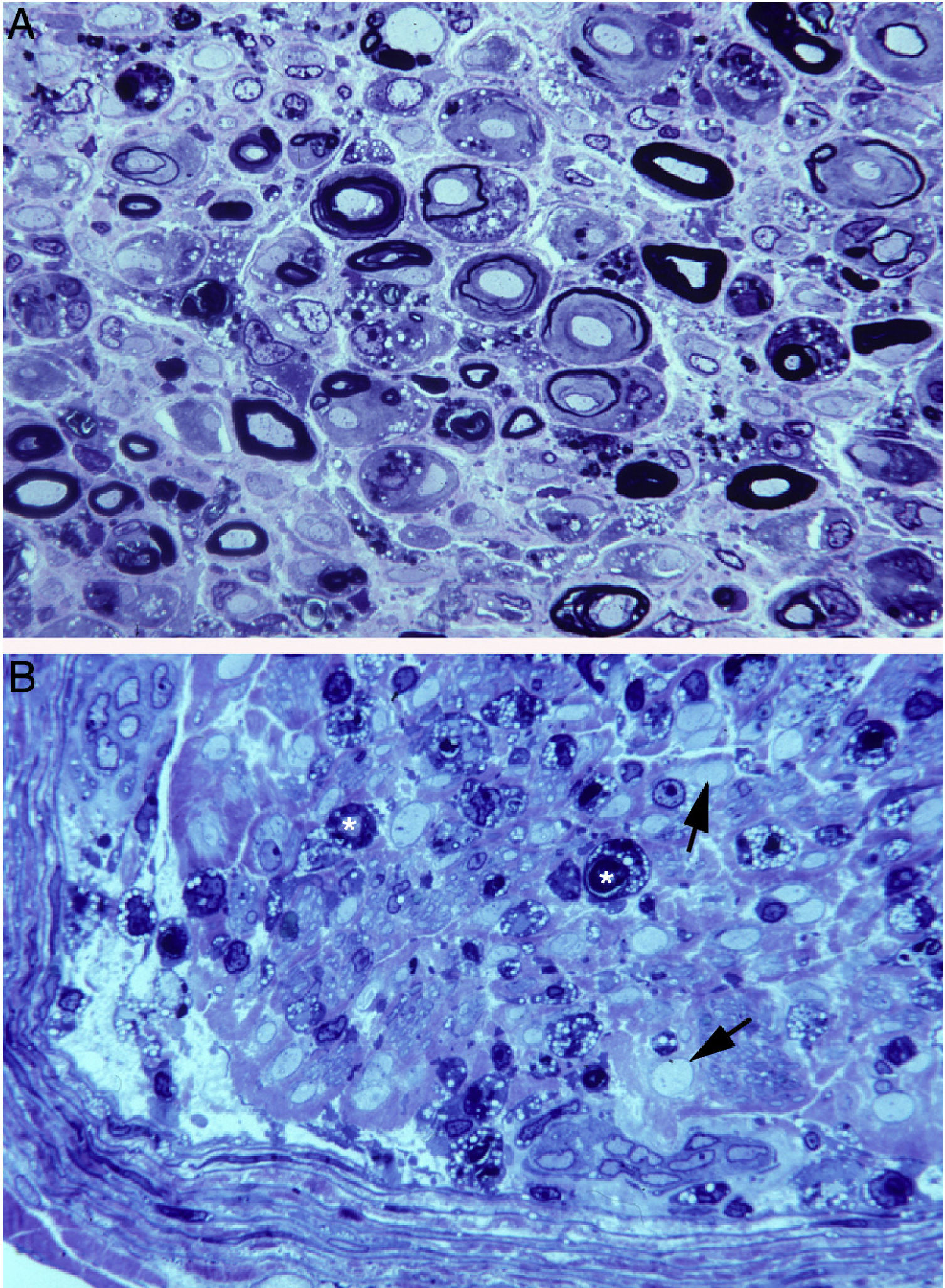
In a clinicopathological study of a fatal case of pure motor and axonal GBS, semithin and ultrathin sections of the ventral and dorsal L5 roots revealed lesions selectively involving the ventral root, presenting a combination of macrophage-associated demyelination and axonal degeneration (Fig. 4A).38 The teased fibre study of the L5 ventral root detected unequivocal signs of primary demyelination,22,39–41 which led us to propose that the case be classified as AIDP with secondary axonal degeneration. We argued that our findings from the semithin sections were compatible with those described by Feasby et al.37 (Fig. 4A and B), and that if those authors had performed a teased fibre study of the nerve root, they may also have detected signs of primary demyelination.
 and figure 2 from Feasby et al.37 (B). Both images show semithin transverse sections of lumbar spinal roots. A) The original annotations are maintained. We observe numerous lipid-laden endoneurial macrophages, sometimes encircling fibres with myelin breakdown (white asterisks) or inside completely destructured nerves. The image also shows clusters of regeneration containing either non-myelinated axons (arrowheads), non-myelinated and thinly myelinated axons (small arrows), and occasional larger fibres with demyelinated or remyelinated axons (large arrows). In the centre of the image is a fibre with vesiculovacuolar dissolution of myelin (black asterisk). This lesion is better documented in figure 6 of Berciano et al.,38 which shows the variety of lesions observed in a teased fibre study. Toluidine blue stain; scale bar: 19μm. B) The caption to the original figure, without annotations, reads: “transverse section showing severe axonal degeneration.” We agree with the authors’ interpretation, as there are indeed numerous axons showing myelin breakdown (white asterisks), which is indicative of acute axonal degeneration.22,39 Note also the presence of fibres with inappropriately thin myelin sheaths (arrowheads) and the fibres exhibiting vacuolar myelin degeneration (black arrows), suggesting de-remyelination; and the clusters of regeneration. Mononuclear inflammatory cells seem to be present in the endoneurial interstitium; the presence of these cells would be better assessed by immunocytochemical techniques.22,39 Although the vesicular dissolution of myelin may have occurred post mortem,40 it was only observed in the lesioned L5 ventral root (and not in the dorsal root, which was spared) in our sample; this supports its morphological value as a sign of demyelination.39 Furthermore, vesicular myelin degeneration is the earliest sign in AIDP, presenting even before the appearance of macrophages in the nerve; this suggests that it is the expression of membrane attack complex formation due to activation of the complement cascade.41 Scale bar: 20μm.Reproduced with permission from Brain.")
Composition of figure 3 from Berciano et al.38 (A) and figure 2 from Feasby et al.37 (B). Both images show semithin transverse sections of lumbar spinal roots.
A) The original annotations are maintained. We observe numerous lipid-laden endoneurial macrophages, sometimes encircling fibres with myelin breakdown (white asterisks) or inside completely destructured nerves. The image also shows clusters of regeneration containing either non-myelinated axons (arrowheads), non-myelinated and thinly myelinated axons (small arrows), and occasional larger fibres with demyelinated or remyelinated axons (large arrows). In the centre of the image is a fibre with vesiculovacuolar dissolution of myelin (black asterisk). This lesion is better documented in figure 6 of Berciano et al.,38 which shows the variety of lesions observed in a teased fibre study. Toluidine blue stain; scale bar: 19μm.
B) The caption to the original figure, without annotations, reads: “transverse section showing severe axonal degeneration.” We agree with the authors’ interpretation, as there are indeed numerous axons showing myelin breakdown (white asterisks), which is indicative of acute axonal degeneration.22,39 Note also the presence of fibres with inappropriately thin myelin sheaths (arrowheads) and the fibres exhibiting vacuolar myelin degeneration (black arrows), suggesting de-remyelination; and the clusters of regeneration. Mononuclear inflammatory cells seem to be present in the endoneurial interstitium; the presence of these cells would be better assessed by immunocytochemical techniques.22,39 Although the vesicular dissolution of myelin may have occurred post mortem,40 it was only observed in the lesioned L5 ventral root (and not in the dorsal root, which was spared) in our sample; this supports its morphological value as a sign of demyelination.39 Furthermore, vesicular myelin degeneration is the earliest sign in AIDP, presenting even before the appearance of macrophages in the nerve; this suggests that it is the expression of membrane attack complex formation due to activation of the complement cascade.41 Scale bar: 20μm.Reproduced with permission from Brain.
Our study38 was met with controversy, with 3 consecutive articles in the “Issues and Opinions” section of the journal Muscle and Nerve (June 1994). One of these noted the difficulty of distinguishing primary axonopathy from axonopathy secondary to inflammatory demyelination in neurophysiological studies42; the authors of the other 2 articles addressed the nosological distinction between axonal GBS43,44 and AMAN.45 In a subsequent fulminant case of motor axonal GBS, the autopsy examination showed demyelination of spinal nerves and roots, sparing the sural and median nerves.46,47
In summary, there is considerable overlap between axonal GBS and AIDP with secondary axonal degeneration; the 2 can sometimes only be distinguished through detailed autopsy studies.
AMAN and its connection with nodo-paranodopathiesIn 1991, McKhann et al.48 described a syndrome of acute flaccid paralysis occurring in summertime in rural areas of northern China. While it was considered a purely motor syndrome, many patients presented pain and meningeal signs. Findings from the first 10 fatal cases of acute flaccid paralysis identified 3 main patterns: 1) 5 cases presenting Wallerian degeneration of the ventral spinal roots and peripheral nerve motor fibres; 2) 3 cases with inflammatory demyelination; and 3) 2 cases without pathogenic lesions.49 The term AMAN was coined to refer to the axonal form. With great precision, the authors described how lesions in AMAN initially appear in the proximal or intermediate section of the ventral root, and increase distally to the point at which the root exits through the dura, where degeneration affects 80% of fibres. Patient serum samples collected in the first 10 days of progression displayed antibodies targeting Campylobacter jejuni in 90% of cases; this pathogenic association had previously been described a decade earlier.50
In a subsequent series of studies, Prof Jack Griffin’s research group completed the histological and immunopathological description of acute flaccid paralysis, performing an additional 12 autopsy studies.41,51–55 Once more, the researchers distinguished 3 basic neuropathological patterns: axonal in 6 cases (3 AMAN and 3 AMSAN), AIDP in 3, and minimal changes in 3 cases. They proposed that AMAN was a new syndrome caused by an autoimmune attack against the nodal axolemma of motor fibres, mediated by antiganglioside antibodies and complement activation.54 Over the last 2 decades, the pathogenic relationship between ganglioside and GBS has been an important area of research (1006 search results on PubMed as of 27 April 2018); it has recently been addressed in several reviews.3,9,56–58
Advances in the understanding of the molecular architecture of nerve fibres and the discovery of nodal and paranodal target antigens59 has led to the emergence of a new pathophysiology of autoimmune neuropathies, and the concept of nodo-paranodopathies.60–64 In AMAN, the autoimmune attack against epitopes at nodes of Ranvier has the following consequences: 1) alteration of sodium-gated channels, which may be associated with reversible conduction failure without accompanying demyelination, in which case consecutive neurophysiological studies should be performed; 2) macrophage invasion of the node region, potentially causing dysfunction of paranodal axo-glial junctions with disruption of the paranodal myelin; and 3) massive Ca++ influx into the axoplasm with active axonal degeneration when the membrane attack complex reaches a critical level.
The concept of nodo-paranodopathies enables novel pathogenic perspectives, which are of great importance when interpreting the neurophysiological alterations occurring in GBS.
“Pure” motor Guillain-Barré syndrome: exclusively a motor and axonal condition?Since the original description of the disease,65 GBS has been known to manifest with mainly motor symptoms.
In 27 of their sample of 147 patients (18%), Visser et al.66 observed almost exclusively motor symptoms with neurophysiological evidence of axonal involvement. Compared to the rest of the patients in their series, this motor subgroup also presented several specific characteristics; therefore, this entity was considered to be similar to AMAN, despite not showing seasonal variation in epidemiology. Since this influential study, the terms pure motor GBS, axonal GBS, and AMAN were often used interchangeably.
In a series of 369 patients with GBS, Hadden et al.67 report 53 cases (16%) of “pure” motor GBS, classified as follows: 1) 27 demyelinating (10% of all cases of demyelinating GBS); 2) 5 axonal (50% of all cases of axonal GBS); and 3) 21 with inexcitability, or unclear or normal neurophysiological findings.
Therefore, while the “pure” motor subtype is more frequent among patients with axonal GBS, it may also present in those with AIDP, including in autopsy-confirmed cases (see above).33,46,47
Pathogenic determinism of the epi-perineurium in lesion causation in Guillain-Barré syndromeAs we have seen, P2-EAN rats show different lesions in the spinal roots and in the sciatic nerve when the immunogen dose is increased.20,21 We have also discussed the predominance of spinal nerve and root involvement in autopsy studies of patients who died early after onset of GBS. This is an opportune moment to consider the question of how a proximal demyelinating neuropathy could be replaced by axonal involvement in more distal nerve trunks. We attempted to solve this puzzle through clinico-pathological studies of GBS.
Consecutive neurophysiological studies (at 3, 10, and 17 days after onset) of a 67-year-old patient with fulminant GBS revealed loss of nerve excitability.68 The histological study revealed the lesions illustrated in Fig. 5, which essentially constitute massive demyelination of the spinal roots and a combination of demyelination and axonal degeneration of more distal nerve trunks. Therefore, these findings accurately reproduce the different involvement of spinal roots and peripheral nerve trunks described in P2-EAN.20,21 In the light of these data and the microscopic anatomy of the spinal cord and spinal nerves and roots (Fig. 6),6,69 we considered it necessary to perform histological studies of the nerve roots, the ventral rami of spinal nerves, and more distal nerve trunks. Given the relatively elastic arachnoid covering of the nerve roots, the initial radicular inflammatory demyelination may be accommodated without an increase in endoneurial pressure. From the subarachnoid angle, the dura mater is replaced by epineurium and the arachnoid mater by the perineurium, which ensheath the peripheral nerve trunks until their motor and sensory terminals. Given that the epi-perineurium is less compliant than the arachnoid mater, we also supposed that critical endoneurial inflammation in early stages of GBS may be accommodated only by increased endoneurial pressure, potentially compromising transperineurial blood flow and ultimately causing endoneurial ischaemia and nerve conduction failure (Fig. 6).
 Semithin L5 ventral root section presenting massive demyelination with numerous lipid-laden macrophages. B) Semithin section from the femoral nerve, presenting numerous fibres with myelin breakdown (asterisks), indicating active axonal degeneration, and demyelinated axons (arrows). Once more, note the presence of lipid-laden macrophages, often encircling fibres exhibiting myelin breakdown. Toluidine blue stain; ×630 before reduction.")
Pathology of fulminant GBS with early nerve inexcitability.69
A) Semithin L5 ventral root section presenting massive demyelination with numerous lipid-laden macrophages.
B) Semithin section from the femoral nerve, presenting numerous fibres with myelin breakdown (asterisks), indicating active axonal degeneration, and demyelinated axons (arrows). Once more, note the presence of lipid-laden macrophages, often encircling fibres exhibiting myelin breakdown. Toluidine blue stain; ×630 before reduction.
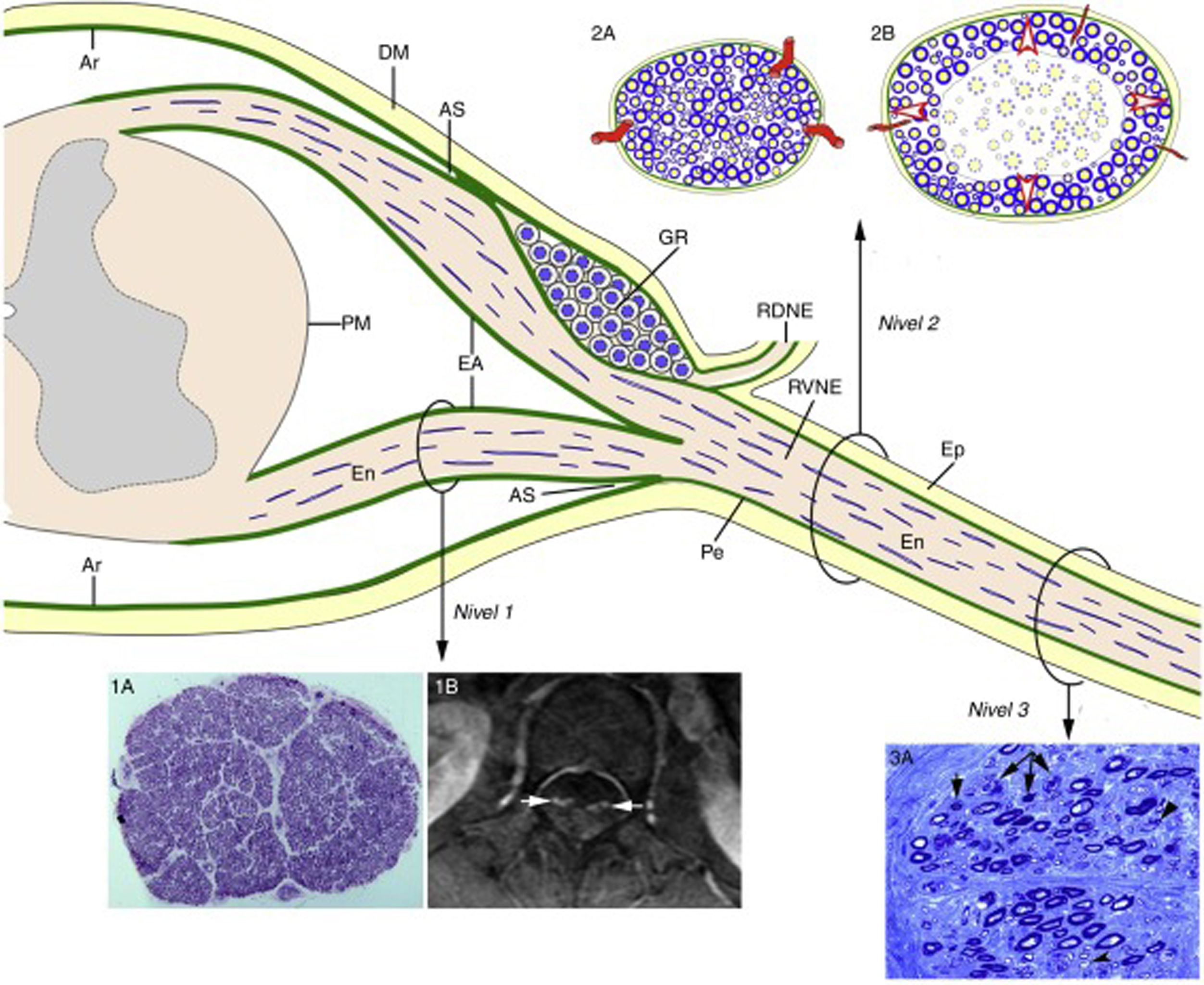
, the epineurium (Ep) is in continuity with the dura mater (DM). The endoneurium (En) of the peripheral nerve trunks extends through the spinal roots until their junction with the spinal cord. At the SA, most of the perineurium (Pe) joins the subdural arachnoid (Ar), with some layers forming the root sheath (RS). The Ar of the spinal roots is continuous with the pia mater (PM) at their emergence from the spinal cord. Immediately after the spinal ganglion (SG), in the SA, the ventral and dorsal roots join to form the spinal nerve, which emerges from the vertebral foramen and splits into the dorsal and the ventral rami (DRSN and VRSN, respectively). Therefore, the arachnoid sheath of the intrathecal spinal roots is elastic, whereas the spinal nerves and more distal nerve trunks are ensheathed in epineurium, which is relatively inelastic. The proximo-distal inflammatory lesions observed in early GBS are illustrated for a lumbar ventral root (Level 1), a spinal nerve (Level 2), and the sciatic nerve (Level 3). The image for Level 1 shows a complete semithin transverse section of the L5 root of a patient with fatal GBS. The density of myelinated fibres is preserved (1A), although the inflammatory lesions visible at greater magnification (not shown) may explain the increased cross-sectional area, enlargement, and the contrast enhancement on MRI sequences (1B, arrows). The diagrams for Level 2 depict the following: a) normal spinal nerve anatomy, usually monofascicular, with transperineurial vessels and epi-perineurial covering (2A), explaining the normal appearance in ultrasound studies, with a round or oval hypoechoic structure surrounded by a hyperechoic rim (see text below); and b) in early GBS, endoneurial inflammatory oedema may cause a critical increase in endoneurial pressure in the spinal nerves, stretching the epi-perineurium beyond the limit of its compliance and constricting the transperineurial vessels; this would result in endoneurial ischaemia, here involving the centre of the fascicle (2B). The image for Level 3 shows a semithin section of the sciatic nerve from a patient with fatal AIDP, displaying several fibres with axonal degeneration (myelin breakdown, arrows), which in this case is secondary to more proximal inflammatory demyelinating lesions. Note also the presence of remyelinated fibres (arrowheads) and lipid-laden macrophages. Were we not aware of the proximal demyelination, it would have been very difficult to accurately interpret the pathogenic role of the florid axonal degeneration observed. This diagram is inspired by figures 2-6 of Berthold et al.68")
Diagram of the microscopic anatomy of the spinal cord, spinal roots, and spinal nerves, taken from Berciano et al.16 From the subarachnoid angle (SA), the epineurium (Ep) is in continuity with the dura mater (DM). The endoneurium (En) of the peripheral nerve trunks extends through the spinal roots until their junction with the spinal cord. At the SA, most of the perineurium (Pe) joins the subdural arachnoid (Ar), with some layers forming the root sheath (RS). The Ar of the spinal roots is continuous with the pia mater (PM) at their emergence from the spinal cord. Immediately after the spinal ganglion (SG), in the SA, the ventral and dorsal roots join to form the spinal nerve, which emerges from the vertebral foramen and splits into the dorsal and the ventral rami (DRSN and VRSN, respectively). Therefore, the arachnoid sheath of the intrathecal spinal roots is elastic, whereas the spinal nerves and more distal nerve trunks are ensheathed in epineurium, which is relatively inelastic. The proximo-distal inflammatory lesions observed in early GBS are illustrated for a lumbar ventral root (Level 1), a spinal nerve (Level 2), and the sciatic nerve (Level 3). The image for Level 1 shows a complete semithin transverse section of the L5 root of a patient with fatal GBS. The density of myelinated fibres is preserved (1A), although the inflammatory lesions visible at greater magnification (not shown) may explain the increased cross-sectional area, enlargement, and the contrast enhancement on MRI sequences (1B, arrows). The diagrams for Level 2 depict the following: a) normal spinal nerve anatomy, usually monofascicular, with transperineurial vessels and epi-perineurial covering (2A), explaining the normal appearance in ultrasound studies, with a round or oval hypoechoic structure surrounded by a hyperechoic rim (see text below); and b) in early GBS, endoneurial inflammatory oedema may cause a critical increase in endoneurial pressure in the spinal nerves, stretching the epi-perineurium beyond the limit of its compliance and constricting the transperineurial vessels; this would result in endoneurial ischaemia, here involving the centre of the fascicle (2B). The image for Level 3 shows a semithin section of the sciatic nerve from a patient with fatal AIDP, displaying several fibres with axonal degeneration (myelin breakdown, arrows), which in this case is secondary to more proximal inflammatory demyelinating lesions. Note also the presence of remyelinated fibres (arrowheads) and lipid-laden macrophages. Were we not aware of the proximal demyelination, it would have been very difficult to accurately interpret the pathogenic role of the florid axonal degeneration observed. This diagram is inspired by figures 2-6 of Berthold et al.68
In 3 subsequent clinico-pathological studies, we analysed whether the radicular inflammatory demyelinating process is modified as the spinal root transitions into the spinal nerve.15,70,71 A dramatic change in lesion pattern was observed between the L5 ventral root and the ventral ramus of the fifth spinal nerve (Figs. 2 and 3). Similar sequential changes were observed in both of the other patients with AIDP, who initially presented an axonal neurophysiological pattern. Fig. 7 shows how nerve trunks with epi-perineurium may display wedge-shaped or centrofascicular areas of pronounced reduction in the number of large myelinated fibres, suggesting the presence of endoneurial ischaemia.22
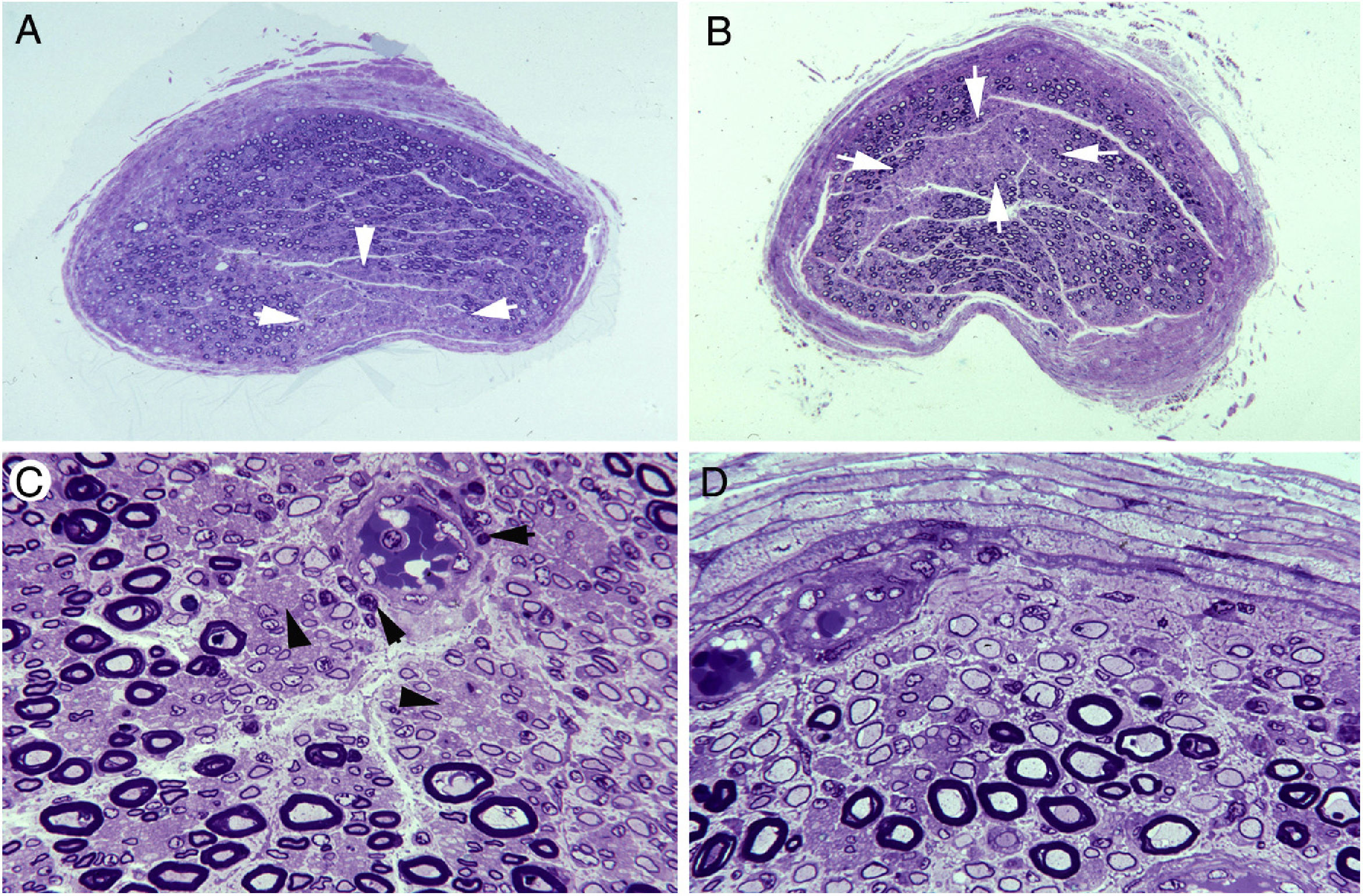
 Semithin transverse section of the third lumbar nerve, showing a wedge-shaped area (arrows) of pronounced loss of myelinated fibres. Toluidine blue stain; ×62 before reduction. B) Semithin transverse section of the lumbosacral trunk, with the centrofascicular area presenting loss of myelinated fibres (arrows). Toluidine blue stain; ×62 before reduction. Note the diffuse reduction in the density of myelinated fibres in both A and B. C) Detailed view of the lumbosacral trunk, showing marked loss of large myelinated fibres; small, thinly myelinated axons; preserved unmyelinated axons (arrowheads); and numerous mononuclear inflammatory cells, some with perivascular distribution (arrows). Toluidine blue stain; ×375 before reduction. D) This subperineurial region of the lumbosacral trunk displays numerous de-remyelinated fibres and mononuclear cells. The extensive de-remyelination explains, to a great extent, the apparent loss of myelinated fibres in A and B. Toluidine blue stain; ×475 before reduction.")
Ischaemic lesions to proximal nerve trunks in AIDP.70
A) Semithin transverse section of the third lumbar nerve, showing a wedge-shaped area (arrows) of pronounced loss of myelinated fibres. Toluidine blue stain; ×62 before reduction.
B) Semithin transverse section of the lumbosacral trunk, with the centrofascicular area presenting loss of myelinated fibres (arrows). Toluidine blue stain; ×62 before reduction. Note the diffuse reduction in the density of myelinated fibres in both A and B.
C) Detailed view of the lumbosacral trunk, showing marked loss of large myelinated fibres; small, thinly myelinated axons; preserved unmyelinated axons (arrowheads); and numerous mononuclear inflammatory cells, some with perivascular distribution (arrows). Toluidine blue stain; ×375 before reduction.
D) This subperineurial region of the lumbosacral trunk displays numerous de-remyelinated fibres and mononuclear cells. The extensive de-remyelination explains, to a great extent, the apparent loss of myelinated fibres in A and B. Toluidine blue stain; ×475 before reduction.
To summarise, the transition from spinal root to spinal nerve involves a dramatic change in early AIDP, with the appearance of endoneurial ischaemic lesions, indicating the pathogenic relevance of inflammatory oedema in nerve trunks with epi-perineurium.
Neurophysiology in the early stages of Guillain-Barré syndromeNerve conduction studies are essential not only for establishing whether the paralytic syndrome is neuropathic, but also to ascertain the specific subtype of GBS.3–6,56 In the first days of progression, neurophysiological studies often return findings not indicative of demyelination or axonal degeneration, such as F-wave or H-reflex alterations.2,72–78 In the first 4 days of progression, the sensitivity of these studies for detecting AIDP ranges from 19% to 63%, depending on the electrodiagnostic criteria used.75
Kurt Incesu et al.79 applied a lumbar root stimulation technique in 15 patients with early GBS, reporting reduced M-response amplitude in all cases; conventional neurophysiological studies, in contrast, returned normal or non-diagnostic results in 6 patients (40%). Temuçin and Nurlu80 studied 12 patients with early GBS, and describe significant prolongation of motor root conduction time in 83% of cases. It has been suggested that alterations in cauda equina conduction time are frequent in patients with AIDP, but not among those with AMAN.81 This finding appears to contradict the results described by Sevy et al.,82 who used a triple stimulation technique in 6 patients with early AMAN in whom conventional neurophysiological findings were non-diagnostic. Proximal conduction blocks were detected at the emergence of the ventral ramus of the spinal nerve through the vertebral foramen. In another study including 13 patients with early GBS, stimulation at the Erb point with recording in the median and cubital nerves detected alterations in compound muscle action potentials in 10 patients (77%).83
The results of all the neurophysiological studies mentioned indicate the considerable pathogenic relevance of proximal motor conduction block in the early stages of GBS; this is perfectly consistent with the lesion topography observed both in AIPD and in AMAN.
Imaging studies in early Guillain-Barré syndromeSpinal magnetic resonance imaging (MRI) and ultrasound of the nerve trunks are the imaging techniques of choice in early GBS. Let us analyse each technique separately.
Spinal magnetic resonance imagingIn many cases, a spinal MRI scan is performed at admission to establish a differential diagnosis between acute myelopathy and ascending paralysis due to GBS.84 Various series38,85–89 of patients with GBS report that contrast enhancement of the spinal roots is detected almost constantly (reviewed in Berciano et al.16). It should be noted that this enhancement may be circumscribed to the ventral roots in pure motor GBS89 and in paraparetic and axonal forms.90–92
Nerve ultrasoundNerve ultrasound is currently a routine diagnostic technique for studying PNS conditions.93 In our series of 6 patients with early GBS (4 AIDP and 2 AMSAN), the main alterations observed in ultrasound studies involved the ventral rami of the cervical nerves, and consisted of enlarged cross-sectional area, loss of the hyperechoic epineurial rim, or both (Fig. 8).15,94 Only 8.8% of peripheral nerve trunk ultrasound studies detected inflammatory alterations, with the median nerve most commonly affected.
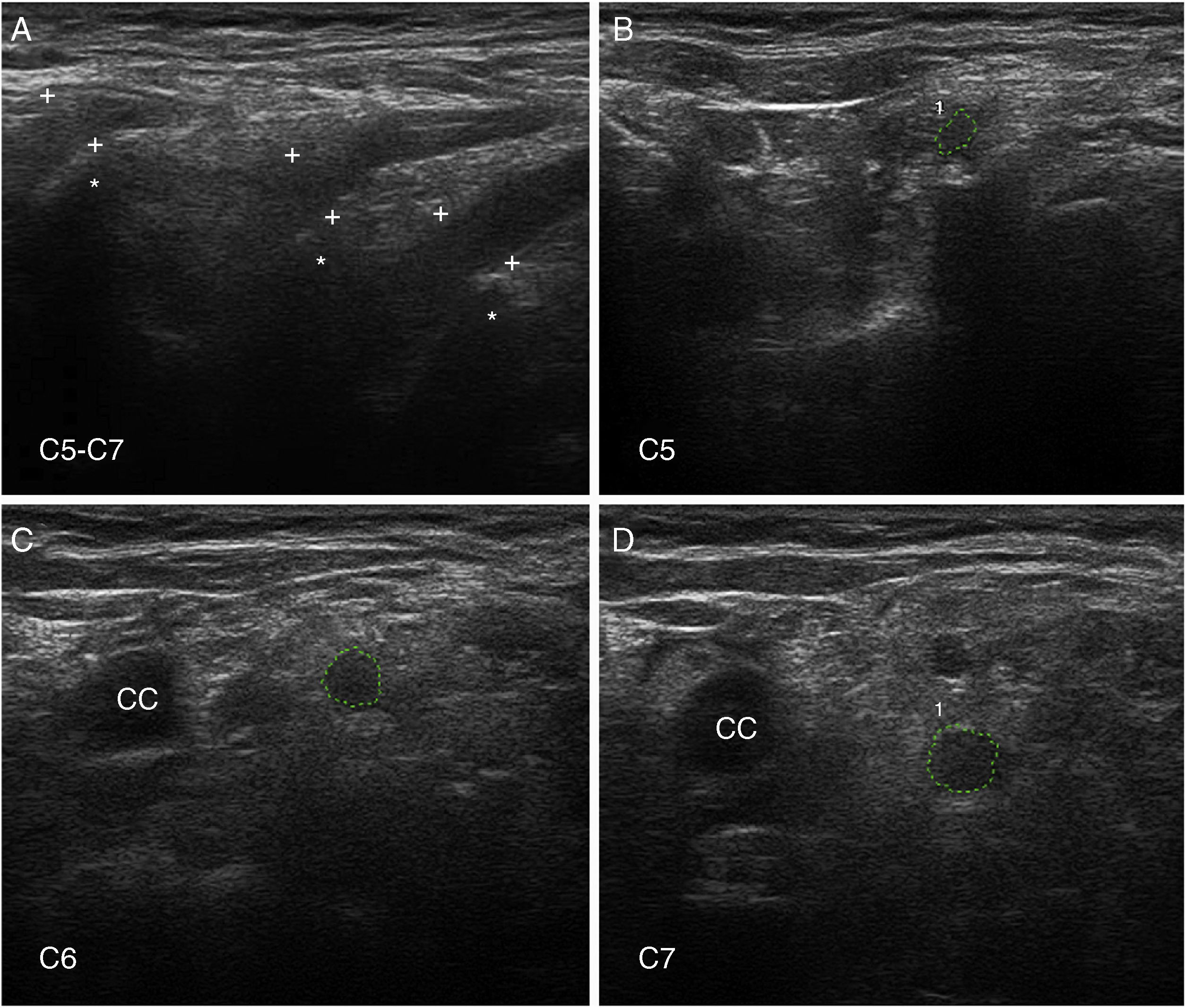
; figures and S1 from Gallardo et al.15 show histology findings from the sixth cervical nerve. A) Sagittal ultrasound image showing blurring of the epineurial covering of all 3 nerves scanned (callipers). The asterisks indicate the transverse processes. B-D) Short-axis ultrasound images of the ventral rami of nerves C5-C7 (dotted green tracings). The nerves show significantly enlarged cross-sectional areas. Note the lack of a hyperechoic epineurial rim; this may be compared against the images published in the normative study by Haun et al.94 The endoneurial inflammatory oedema (see Fig. 2C-D and 3) explains the enlarged cross-sectional area, while the epi-perineurial inflammatory process (see Fig. 2E) would account for the blurring of the hyperechoic epineurial rim.")
Ultrasound image of cervical nerves from a patient with AIDP (the same patient as Figs. 2 and 3); figures and S1 from Gallardo et al.15 show histology findings from the sixth cervical nerve.
A) Sagittal ultrasound image showing blurring of the epineurial covering of all 3 nerves scanned (callipers). The asterisks indicate the transverse processes.
B-D) Short-axis ultrasound images of the ventral rami of nerves C5-C7 (dotted green tracings). The nerves show significantly enlarged cross-sectional areas. Note the lack of a hyperechoic epineurial rim; this may be compared against the images published in the normative study by Haun et al.94 The endoneurial inflammatory oedema (see Fig. 2C-D and 3) explains the enlarged cross-sectional area, while the epi-perineurial inflammatory process (see Fig. 2E) would account for the blurring of the hyperechoic epineurial rim.
In contrast to our findings, other authors do report enlarged cross-sectional areas of peripheral nerve trunks in early GBS, although only regressive oedema of nerves C5 and C6 seems to correlate with clinical improvement.95–97 Given the importance of the examiner’s skill in nerve ultrasound studies, further research is needed to more precisely determine the technique’s usefulness as a diagnostic test for GBS.98,99
Diagnostic considerationsThe involvement mainly of the spinal nerves in very early AIDP (≤ 4 days) explains why conventional nerve conduction studies may return normal or non-specific findings, even in patients with severe paralysis. In the subsequent progression, there are 2 main possibilities16,90: 1) massive endoneurial ischaemia involving the spinal nerves, resulting in axonopathy, with the initial neurophysiological pattern changing to one of axonal degeneration; and 2) demyelinating lesions to nerve trunks, with neurophysiological studies indicating a demyelinating pattern.
In the early stage of AMAN, inflammatory oedema of proximal nerve trunks may have comparable effects to those of AIDP. Antiganglioside antibodies may also play a pathogenic role, causing non-demyelinating reversible conduction failure of proximal, intermediate, or distal nerve trunks. In AMAN and AMSAN, the progression from axonal dysfunction to Wallerian degeneration depends both on the impact of the initial inflammatory oedema and on the degree of antiganglioside antibody–induced disruption of the nodal/paranodal axolemma.60–62 In any case, consecutive neurophysiological studies are invaluable in assessing non-demyelinating reversible conduction failure and the progressive axonal degeneration pattern.100
Therapeutic considerationsSpecific pharmacological treatment of GBS mainly consists in the use of intravenous immunoglobulins and plasmapheresis.5,6,56 Treatment with corticosteroids is generally discouraged.5,6,56 However, there are 2 situations that merit separate consideration.
Firstly, corticotherapy may be warranted in fulminant cases of GBS with early nerve trunk inexcitability.69 The pathogenic role of endoneurial oedema in these patients is as significant as that underlying any decompensation of intracranial hypertension.16,101 The administration of pulses of intravenous methylprednisolone or dexamethasone is totally justified in these circumstances. We may also consider in future the use of other anti-oedema treatments.
The second situation is related to the semiology of pain. Nerve trunk pain is a common manifestation of both AIDP and AMAN,102 preceding paralysis in one-third of cases.103–105 Pain has been associated with inflammation of the dorsal spinal roots, which is relevant in AIDP but not AMAN (in which, by definition, the dorsal roots are spared). As we have seen, AMAN pathology systematically involves the ventral roots, extending to the ventral rami of the spinal nerves. The dorsal rami are located adjacent to the ventral rami (Fig. 6). The initial inflammatory oedema of the ventral rami of the spinal nerves may extend to the dorsal rami by 2 mechanisms16: 1) by simple continuation of the inflammatory process, given that these are adjacent structures; or 2) by increased endoneurial pressure on the spinal nerves with endoneurial ischaemia, which would inevitably affect fibres that join the dorsal rami. This inflammatory basis would justify the use of corticosteroids to manage intractable pain in GBS: 13 well-documented cases have been presented of patients with severe, refractory back pain responding well to corticotherapy.16
ConclusionEarly GBS involves inflammatory oedema of proximal nerve trunks, which may lead to nerve conduction failure and active axonal degeneration.
FundingThis study received no specific funding from any public or private organisation.
Conflicts of interestThe author has no conflicts of interest to declare.
This review summarises 3 decades of work on GBS, in which I have benefited from the invaluable assistance of my colleagues. I am particularly grateful to Dr María José Sedano of the neurology department of Hospital Universitario Marqués de Valdecilla (HUMV), Dr Antonio García and Dr Pedro Orizaola of the clinical neurophysiology department of HUMV, Dr Javier Figols and Dr Nuria Terán-Villagrá of the anatomical pathology department of HUMV, Dr Elena Gallardo of the radiodiagnostics department of HUMV, and to Prof Miguel Lafarga and Prof María Teresa Berciano of the Department of Anatomy and Cell Biology at Universidad de Cantabria. I would also like to thank Dr José Gazulla (Hospital Miguel Servet, Zaragoza) for reviewing the manuscript, and Mario Corral (director of the Marquesa de Pelayo library in Santander) for his assistance with the literature search.
Please cite this article as: Berciano J. Patología axonal en la fase precoz del síndrome de Guillain-Barré. Neurología. 2022;37:466–479.
