



La ataxia por déficit de vitamina E (AVED) es un trastorno autosómico recesivo causado por mutaciones en el gen codificante para la proteína transportadora de α-tocoferol (α-TTPA), localizado en el cromosoma 8q12.31. Dichas mutaciones conllevan niveles bajos de vitamina E como resultado de un defecto de su transporte hepático tras una adecuada absorción intestinal2. Clínicamente se traduce en un síndrome cerebeloso con ataxia junto con respuesta cutáneo-plantar (RCP) extensora, arreflexia y déficits sensitivos superponible a la ataxia de Friedreich3,4. No obstante, puede asociar otras manifestaciones neurológicas (distonía, temblor, mioclonus) o extraneurológicas (retinitis pigmentosa, xantelasmas, xantomas tendinosos o alteraciones cardiacas)5.
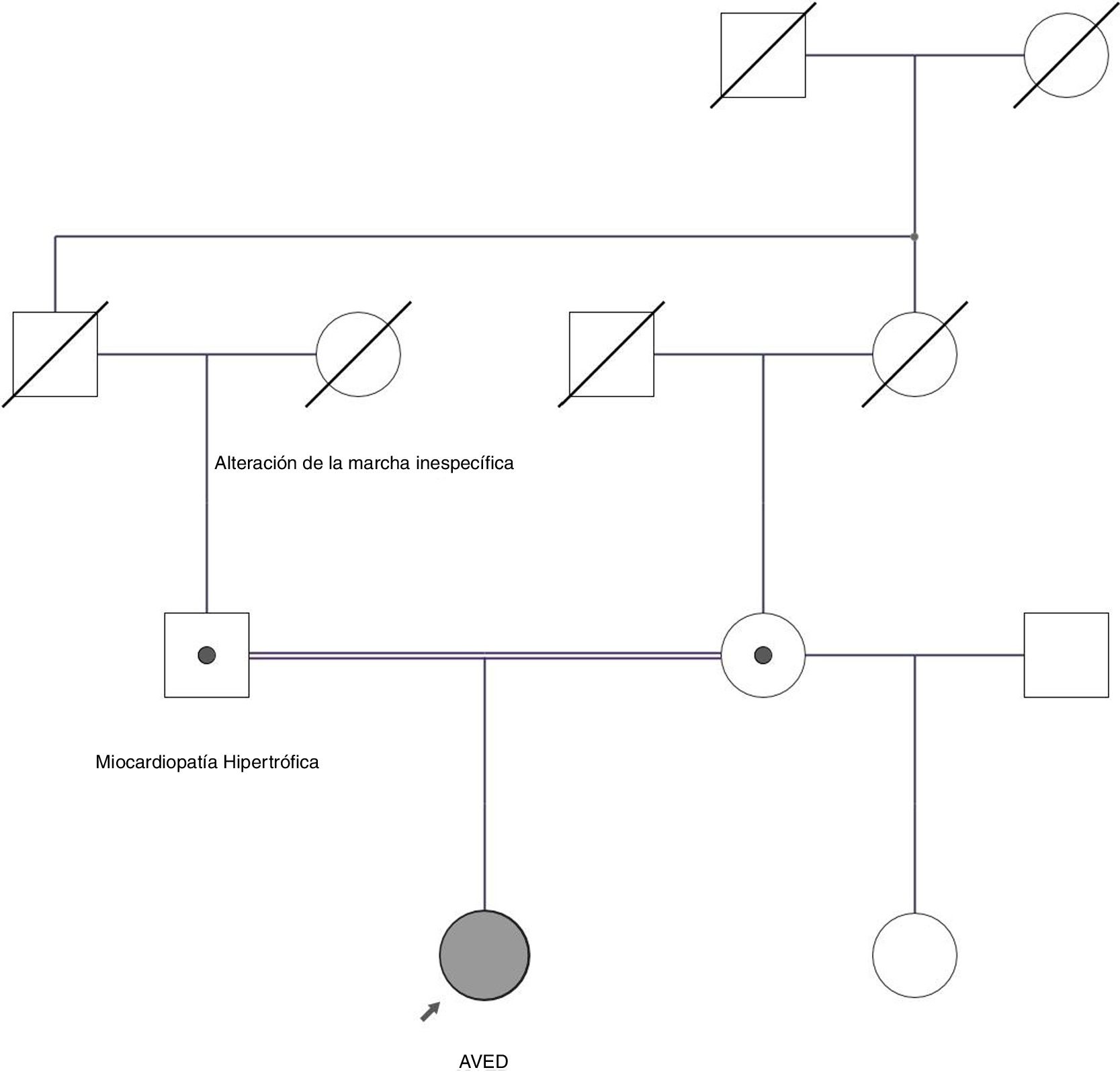
Describimos el caso de una mujer de 27 años con dificultad para el habla y la marcha iniciadas hacia los 8 años. Posteriormente desarrolló una ataxia cerebelosa con caídas. Como antecedentes familiares destacan abuela paterna con cuadro de alteración de la marcha y padres consanguíneos (fig. 1). El padre presentaba una miocardiopatía hipertrófica obstructiva.

Árbol genealógico. Se observa que los padres de la paciente son consanguíneos primos-hermanos, siendo ambos portadores de mutación patógena en heterozigosis. El padre se encuentra afecto de miocardiopatía hipertrófica. La abuela paterna se encontraba afecta de alteración de la marcha inespecífica. Se desconoce el estado de portador de familiares fallecidos.
El examen general evidenció cifoescoliosis y pies cavos. La exploración neurológica objetivó disartria escandida con nistagmo evocado agotable a la mirada extrema bilateral con leve aumento de latencia en los movimientos sacádicos. Existía hipotonía global, dismetría bilateral, disdiadococinesia, ataxia troncular con signo de Romberg, marcha inestable de base amplia con paso irregular y tándem dificultoso. Destacaba una arreflexia generalizada junto con RCP extensor bilateral, hipopalestesia en extremidades inferiores. La resonancia magnética craneal mostró un platibasia. La electromiografía detectó una marcada alteración de la vía cordonal posterior y la vía piramidal tanto para extremidades superiores como inferiores. El ecocardiograma transtorácico resultó normal.
Ante el cuadro de ataxia progresiva con arreflexia y RCP extensor bilateral se consideró como primera opción diagnóstica una ataxia de Friedreich, aunque el análisis del gen de la frataxina fue normal. El estudio bioquímico confirmó la deficiencia de vitamina E siendo la concentración de α-tocoferol de 4,72μmol/l (18,6-46,2μmol/l). El resto de datos de laboratorio resultaron normales. El diagnóstico se confirmó mediante secuenciación del gen TTPA, siendo la paciente portadora homocigota del cambio patogénico c.513_514insTT. El estudio de ambos progenitores mostró niveles de α-tocoferol normales en la madre y disminuidos en el padre, quien presentó una concentración de 11,61μmol/l. Ambos progenitores resultaron portadores heterocigotos c.513_514insTT6–8. Se realizó un estudio genético extendido respecto la miocardiopatía, siendo el padre portador heterocigoto del cambio c.4288T>G en el MYH6 gen. Dicho gen codifica la cadena pesada de la subunidad α de la miosina cardiaca, expresada principalmente durante el período embrionario. Alteraciones en este gen se han implicado en un amplio espectro de enfermedades cardiacas incluyendo cardiomiopatías dilatadas e hipertróficas9 entre ellas la cardiomiopatía hipertrófica familiar 14 (OMIM #613251). c.4288T>G implica la sustitución de una leucina en la posición 1.430 por una valina. El estudio bioinformático considera esta variante de significado incierto.
La paciente inició tratamiento con vitamina E parenteral a dosis de 800mg/día, sin embargo, los niveles de vitamina E plasmáticos permanecieron bajos, procediéndose a doblar la dosis. Clínicamente ha permanecido estable.
La vitamina E es una molécula liposoluble antioxidante con acción protectora frente a enfermedades relacionadas con estrés oxidativo como las enfermedades cardiovasculares, neurodegenerativas o neoplásicas, gracias a su capacidad de eliminación de especies reactivas de oxígeno10. La deficiencia de vitamina E comporta problemas neurológicos derivados del descontrol en el estrés oxidativo con pérdida neuronal y desmielinización. Además se generan depósitos con productos derivados de la peroxidación lipídica y agregados de lipofuscina, conllevando un transporte axonal deficitario11,12. Estudios post mortem correlacionan el fenotipo atáxico con una atrofia de las neuronas de Purkinje cerebelosas13.
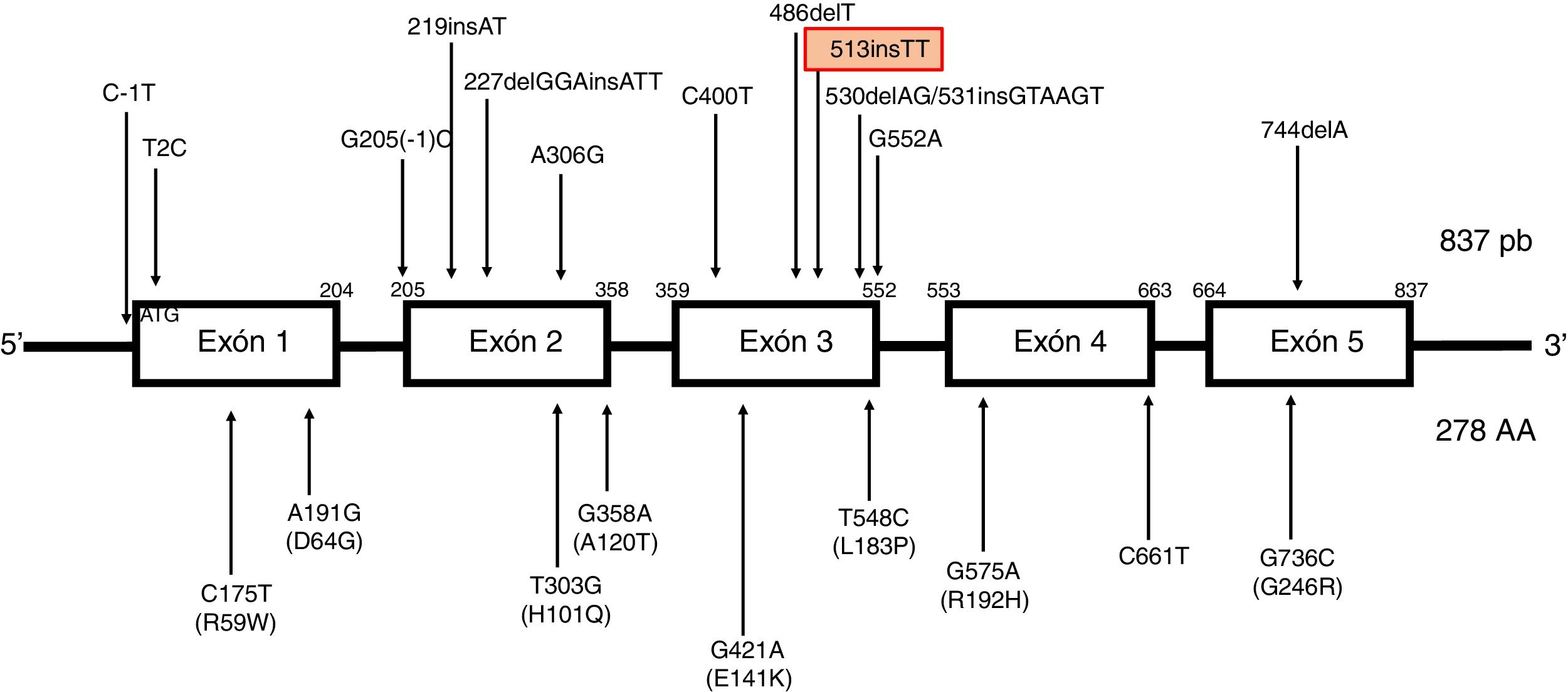
Las mutaciones más frecuentes son el cambio de marco de lectura c.744delA (con alta frecuencia y efecto fundador en poblaciones norteafricanas) y c.513insTT en segundo lugar. Dicha variante patogénica consiste en la inserción de 2 residuos de timina en la posición de nucleótido 513, causando un cambio de marco de lectura y la aparición de un codón de parada prematuro. Se ha descrito en pacientes de origen norte europeo, holandeses, alemanes, daneses e italianos4, asociada predominantemente a clínica cerebelosa (fig. 2).
, sin sentido (nonsense) y de sitio de empalme (splicing site mutations), así como pequeñas deleciones, inserciones e indeleciones (es decir, deleción e inserción simultáneas). AVED muestra una penetrancia casi completa en individuos que son homocigotos o heterocigotos compuestos para una mutación TTPA. Actualmente se han descrito mutaciones en todos los exones. Las mutaciones más frecuentes son el 744delA en el exón 5, predominante en poblaciones del norte de África y la mutación 513insTT en el exón 3, más frecuente en las familias de origen norte de Europa.")
Descripción del gen TTPA. Localizado en el cromosoma 8, consta de 5 exones empalmados uniformemente con un marco de lectura abierto de 834 pares de bases y codifica una proteína citosólica de 278 aminoácidos. TTPA es el único gen descrito asociado con AVED. No se han identificado polimorfismos de un solo nucleótido en la región de codificación de TTPA. Las mutaciones que causan enfermedades de TTPA incluyen mutaciones con cambio de sentido (missense), sin sentido (nonsense) y de sitio de empalme (splicing site mutations), así como pequeñas deleciones, inserciones e indeleciones (es decir, deleción e inserción simultáneas). AVED muestra una penetrancia casi completa en individuos que son homocigotos o heterocigotos compuestos para una mutación TTPA. Actualmente se han descrito mutaciones en todos los exones. Las mutaciones más frecuentes son el 744delA en el exón 5, predominante en poblaciones del norte de África y la mutación 513insTT en el exón 3, más frecuente en las familias de origen norte de Europa.
La presencia de miocardiopatía ha sido reportada en series de casos con una prevalencia entre 19-31%4,14,15, en forma de hipertrofia ventricular izquierda, miocardiopatía dilatada o alteraciones electrocardiográficas. Adicionalmente, se ha reportado en pacientes con AVED el desarrollo de trastornos sistémicos prematuros (como enfermedad vascular aterosclerótica o cardiopatía isquémica y esteatosis hepática) en ausencia de factores de riesgo relevantes, dado que el α-tocoferol podría jugar un papel protector de la función miocárdica al reducir el estrés oxidativo15,16.
No obstante, la presencia de cardiopatía en un portador heterocigoto no ha sido descrita previamente en la literatura. Hipotetizamos que, niveles bajos de α-tocoferol mantenidos de forma sostenida en el tiempo puedan estar en relación a la existencia de afectación cardiaca, ya que tanto los pacientes afectos (homocigotos y heterocigotos compuestos) como aquellos heterocigotos en los cuales presenten niveles bajos de α-tocoferol comparten el factor metabólico principal que conduciría a la existencia de la miocardiopatía descrita en el AVED. Asimismo, cabe destacar que la madre la de la paciente también es heterocigota para la misma mutación. Sin embargo, presenta unos niveles de α-tocoferol dentro de los límites de la normalidad y se encuentra asintomática tanto a nivel neurológico como cardiaco. Este hecho pone de manifiesto la posibilidad de la existencia de otros factores que puedan estar implicados en la relación genotipo-fenotipo en cuanto a los niveles de α-tocoferol. Por otra parte, el estado de portador heterocigoto para múltiples mutaciones recesivas puede representar una situación de vulnerabilidad, actuando en combinación y de esta forma predisponiendo a la expresión fenotípica. En nuestro caso, el déficit de α-tocoferol puede favorecer la expresión fenotípica de la mutación heterocigota en MYH6.
La importancia de la sospecha clínica radica en la realización de un diagnóstico precoz, permitiendo iniciar de forma temprana un tratamiento sustitutivo y así evitar el desarrollo de signos y síntomas incapacitantes17. Por último, sería recomendable el estudio cardiológico en pacientes y portadores.
FinanciaciónEste trabajo no ha sido financiado por ninguna entidad pública o privada.

