



Autosomal dominant hereditary spastic paraplegia (AD-HSP) is a group of neurodegenerative diseases of genetic origin that cause progressive spasticity and weakness in the lower limbs due to degeneration of the corticospinal tract.1 Seventy-nine loci and more than 70 pathogenic variants causing HSP have been described.2 Forty percent of all families with AD-HSP present pathogenic variants of the SPAST gene, located at 2p22.3. This gene, also known as SPG4, contains 17 exons and codes for the protein spastin. Spastin is an ATPase associated with several cellular activities, such as proteolysis, the cell cycle, vesicular transport, peroxisome biogenesis, and mitochondrial functions.3
More than 250 pathogenic variants of different types (deletions, insertions, and base substitutions) have been described in the SPAST gene, and the majority of families described show private mutations. Some of these variants seem to lead to certain peculiarities, such as earlier onset and greater severity in men, cognitive and bladder impairment, or late onset.
The clinical phenotype is indistinguishable between the different mutation mechanisms (nonsense, deletion, reordering, etc.), with haploinsufficiency being the molecular basis of this variant.4
When symptoms manifest during childhood, other diagnoses should be considered, including structural lesions, infections, and metabolic diseases. The pathogenic variants causing HSP of childhood onset occur most frequently in the ATL1 (SPG3A) gene, followed by the SPAST gene.5
We present a new pathogenic variant of the SPAST gene in 3 members of a Spanish family with AD-HSP (Fig. 1).
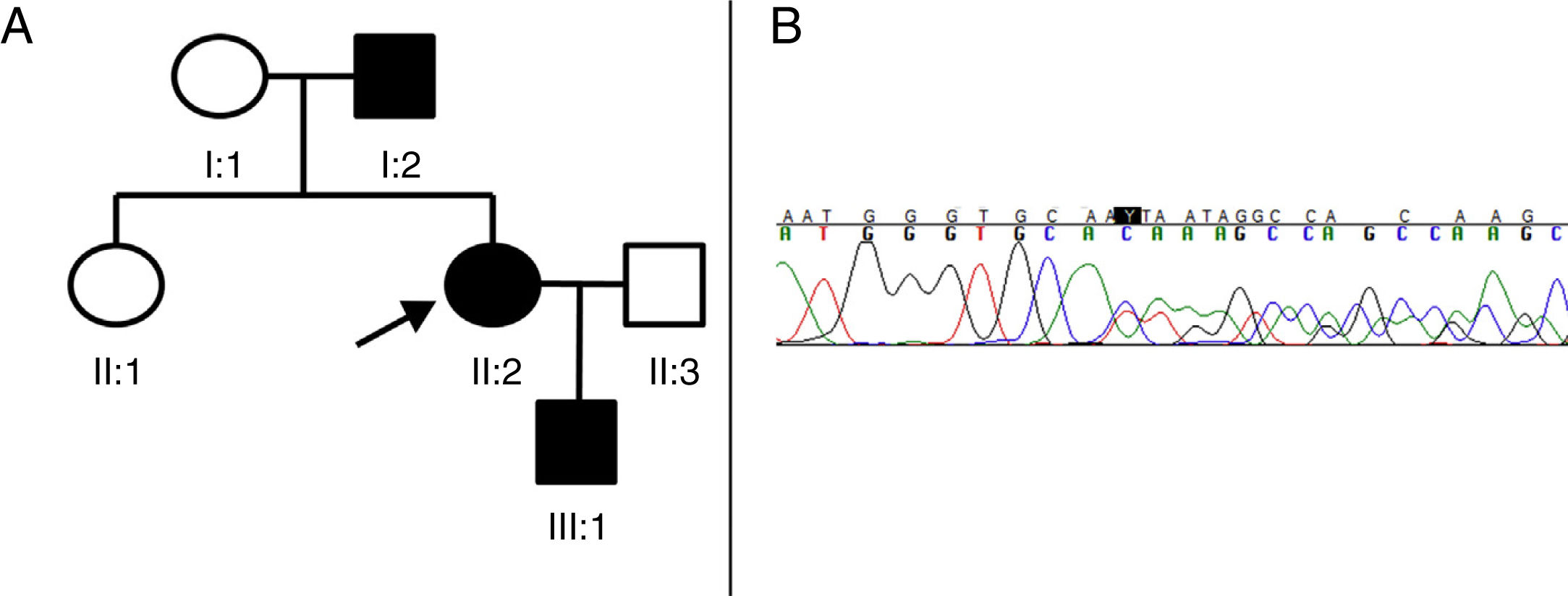
 Pedigree of the family with AD-HSP. The index case (proband) is indicated with an arrow. Squares represent men, circles women, and solid black symbols represent patients with AD-HSP. (B) Segment of sequencing from patient II:2. A heterozygous deletion of 4 nucleotides (TGTC) is detected at position c.1457 in exon 12 of the SPAST gene (c.1457_1460del), causing a premature stop codon (Thr486Ilefs*43).")
(A) Pedigree of the family with AD-HSP. The index case (proband) is indicated with an arrow. Squares represent men, circles women, and solid black symbols represent patients with AD-HSP. (B) Segment of sequencing from patient II:2. A heterozygous deletion of 4 nucleotides (TGTC) is detected at position c.1457 in exon 12 of the SPAST gene (c.1457_1460del), causing a premature stop codon (Thr486Ilefs*43).
Patient II:2 (the proband) is a 41-year-old woman presenting only hyperreflexia and mild spasticity in the lower limbs. Patient I:2 is the father of the index case, and started to develop symptoms at the age of 73, manifesting with progressive weakness and spasticity in the lower limbs, making him unable to walk independently for a period of 3 years. Brain and spinal neuroimaging, cerebrospinal fluid study, and microbiological studies (HIV, syphilis, and HTLV1) revealed no alterations. Finally, patient III:1 is a 4-year-old boy, the son of the index case, who presented patellar tendon hyperreflexia and toe-walking (with no Babinski sign or clonus), which improved with physiotherapy.
As we suspected the possibility of an HSP with a dominant inheritance pattern, we decided to perform sequencing of the SPAST gene. All 3 patients gave informed consent for blood samples to be taken for the genetic study. The 17 exons and adjacent regions of the SPAST gene were amplified by polymerase chain reaction and analysed using single-strand conformation polymorphism analysis by capillary electrophoresis. Sequencing identified a deletion of 4 nucleotides (c.1457_1460del:TGTC) in heterozygosis, causing the substitution of a threonine with an isoleucine and the appearance of a premature stop codon 43 amino acids later (Thr486Ilefs*43) (Fig. 1B). We identified this to be a new variant, classified as pathogenic according to the interpretation guidelines from the American College of Medical Genetics and Genomics,6 as it causes a reading frame shift and codes for a truncated and dysfunctional spastin. This pathogenic variant has not been described in the literature or in the data bases consulted (Exome Aggregation Consortium, Single Nucleotide Polymorphism, and NHLBI Exome Sequencing Project).
In summary, we have identified a new pathogenic variant of the SPAST gene, c.1457_1460del (Thr486Ilefs*43), which is associated with AD-HSP. It is important to highlight, firstly, that while patients may present the same pathogenic variant, phenotype may be heterogeneous, and the absence of symptoms does not rule out the disease; therefore, it should be considered in genetic counselling. Secondly, we should mention the anticipation phenomenon observed in patients II:2 and III:1.
Please cite this article as: Bertran Recasens B, Figueras Aguirre G, Aznar-Lain G, Rubio MA. Nueva variante patogénica en el gen SPAST en una familia española afecta de paraplejía espástica hereditaria. Neurología. 2020;35:340–341.
