



La lipoidoproteinosis (LP), hialinosis cutis et mucosas o enfermedad de Urbach-Wiethe (OMIM 247100) es un raro trastorno autosómico recesivo. El curso es lento y carácter benigno; un total de 2501 a 300 casos2,3 están comunicados. Caracterizado por depósitos intercelulares de material hialino, ácido periódico Schiff positivo en la piel, membranas mucosas y órganos internos4-6.
Es una genodermatosis debida a mutaciones de pérdida de función del gen codificador de la proteína 1 de la matriz extracelular (ECM1) en el cromosoma 1q211,7-9. ECM1 contiene 10 exones con 3 isoformas (ECM1a [la más frecuente], ECM1b y ECM1c), cuyas funciones son todavía mal conocidas. EMC1 se expresa en la dermis, queratinocitos, células endoteliales y los huesos en desarrollo, y está ligada a la diferenciación de los queratinocitos, la regulación de membrana basal, la composición del colágeno y el factor de crecimiento (homeostasis de la piel)1,9.
Las manifestaciones clínicas de la LP son proteicas con variación interindividual5. Tiene una implicación sistémica múltiple, aunque la piel y las mucosas del tracto aéreo-digestivo superior son los principalmente afectados7. Típica, y casi patognomónicamente, se inicia en la infancia con un llanto débil y una voz ronca que es debida a la infiltración laríngea10. Algo más tarde, 3 años edad, la piel sufre una infiltración y engrosamiento difuso, dando lugar a pápulas y cicatrices variceliformes. En el 50-60% de los casos presenta blefarosis moniliforme, a modo de «collar de perlas», esto es casi patognomónico11. En un 50-70% de los casos se producen calcificaciones bilaterales, circunscritas y simétricas en la región medial de los lóbulos temporales, incluyendo los unci hipocampales y el complejo amigdalino visibles en la TC y la RM craneal12, poniendo en evidencia actividad epiléptica, así como alteraciones mnésicas, cambios conducta social y de comportamiento, síntomas paranoicos y retraso mental1,8,12,13.
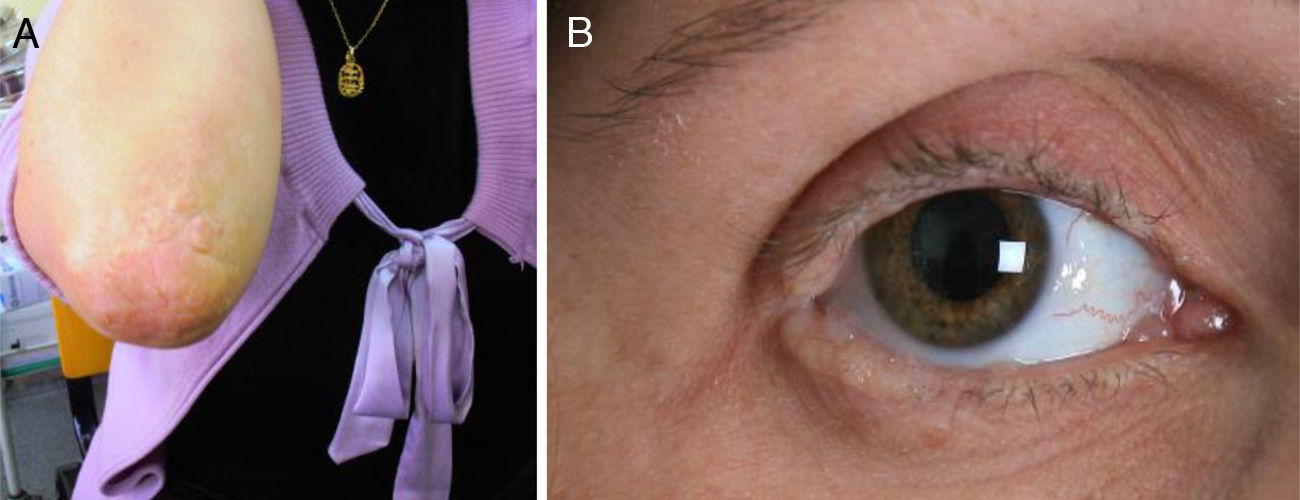
Presentamos el caso de una mujer de 35 años de edad, con padres no consanguíneos, que desde los 9 meses de edad presentaba una voz ronca y disfónica, que venía consultando en nuestro servicio desde 1997 por quejas diversas: dolor de cabeza, pérdida de memoria, «sensación de haber experimentado cosas que no eran verdaderas», «dificultad para reconocer los sitios comunes», mareos, inestabilidad, ansiedad y depresión. Antecedentes de hipotiroidismo. Su exploración mostró una voz disfónica de bajo tono. En superficies de extensión de las articulaciones presentaba múltiples pápulas verrucosas hiperqueratósicas y amarillentas, con aspecto de «empedrado», así como afectación de las mucosas lingual, labial, yugal y blefarosis moniliforme (fig. 1). La exploración neurológica fue normal. La neuropsicológica mostró una leve disminución en la velocidad de procesamiento de la información y leves alteraciones en la memoria episódica y en los procesos de evocación.
 Placas hiperquerátosicas en zonas de extensión en codos y blefarosis morbiliforme.")
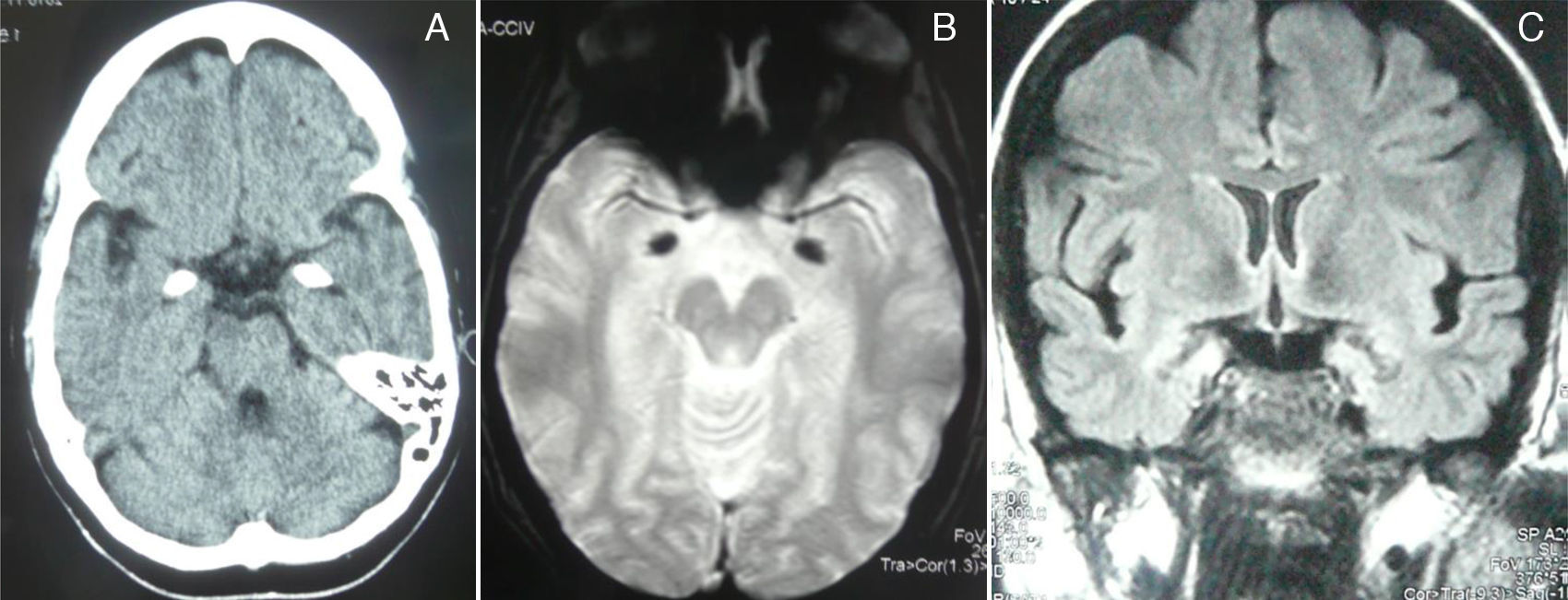
Un completo estudio analítico, hormonal, anticuerpos, inmunidad, serología fue normal, negativo o sin significación clínica. La TC y la RM de cráneo (secuencias de eco-gradiente T2*) mostraron unas lesiones hiperdensas e hipointensas, respectivamente, en regiones amigdalouncales simétricas y bilaterales compatibles con calcificaciones (fig. 2). El electroencefalograma en privación de sueño, normal. Los potenciales evocados cognitivos (P-300) mostraron un retraso en el tiempo de reacción durante el desarrollo de la tarea de Posner, con una latencia normal del componente P3 durante la tarea «oddball».
 TAC s/c y RM craneal secuencia eco gradiente T2* axial y FLAIR coronal: se visualizan hiperdensidades, hiposeñal e hiperseñal, respectivamente, en regiones amigdalouncales simétricas y bilaterales compatibles con calcificaciones.")
La biopsia de piel: cambios dérmicos y epidérmicos con acantosis irregular, hiperqueratosis y deposición de material homogéneo, eosinófilo, hialino, fuertemente PAS+y diastasa resistente alrededor de los vasos sanguíneos dérmicos y estructuras anejas, revelando su naturaleza glucoproteica consistente con LP.
La secuenciación de la región codificante del gen ECM1 mostró una mutación sinsentido en el exón 7 de ECM1, c.1076G>A, que provocaba la aparición de un codón de terminación prematuro, p.Trp359*, que afectaba a la isoforma ECM1a de la proteína 1 de la matriz extracelular.
Son necesarios estudios adicionales para poder establecer una relación genotipo-fenotipo más específica; en nuestro caso, se plantea que la nueva mutación podría dar lugar a un fenotipo cutáneo-mucoso-cerebral más extenso. De la misma manera que las mutaciones fuera del exón 7, como las que afectan a la isoforma ECM1b (que carece de exón 7) se ha asociado a un fenotipo cutaneomucoso (pero no neurológico) más grave14.
En nuestro conocimiento, este es el primer caso de LP con calcificaciones cerebrales confirmadas por análisis genético descrito en la literatura española.
Conflicto de interesesNo existen conflictos de intereses.
La parte genética de este artículo fue previamente publicada en J Clin Neurol. 2014;10:64-8.

