

La hipouricemia se diagnostica cuando los niveles plasmáticos de ácido úrico son menores o iguales a 2,0 mg/dl. El diagnóstico diferencial de la hipouricemia se realiza en función de la excreción fraccional de ácido úrico, y se han identificado varios transportadores y proteínas implicados en el manejo del ión urato en el túbulo proximal. En este artículo se revisan los conocimientos actuales sobre el manejo tubular renal del ácido úrico y las distintas situaciones clínicas asociadas con hipouricemia.
Hypouricemia is defined when a serum urate concentration is less than or equal 2.0mg/dl. Differential diagnosis is made by fractional uric acid excretion with the identification of urate transporters and intracellular proteins involved in the tubular transport of uric acid. This review examines current knowledge on uric acid tubular transport and the various clinical situations of hypouricemia.
INTRODUCCIÓN
La hipouricemia no tiene síntomas reconocidos y, por lo tanto, no requiere tratamiento. Sin embargo, es un signo analítico al que se debe prestar atención dado que se puede asociar con tubulopatías primarias o secundarias y con otras enfermedades subyacentes. Tradicionalmente, los clínicos no hemos dado la misma importancia a la hipouricemia que a la hiperuricemia, quizá por su menor frecuencia de presentación. Sin embargo, hoy en día es cada vez más frecuente la observación de hipouricemia en las consultas de nefrología dado que prestamos asistencia a un mayor número de pacientes con nefropatía diabética y porque cada vez es más frecuente atender a pacientes de diferentes razas y países. Además, desde un punto de vista fisiológico, se han realizado nuevas aportaciones sobre los mecanismos de transporte de ácido úrico en el túbulo proximal renal, por lo que en los próximos años podrían aparecer nuevas terapias destinadas a bloquear o a estimular dichos mecanismos de transporte.
VALORES PLASMÁTICOS Y ELIMINACIÓN URINARIA NORMALES DE ÁCIDO ÚRICO
Las concentraciones séricas de urato son más elevadas en hombres que en mujeres. Así, se define como hiperuricemia, en los primeros, la existencia de unos valores superiores a 7 mg/dl y en las segundas, los que sobrepasan 6 mg/dl. El urato eliminado por la orina constituye aproximadamente el 70% de la producción diaria. El resto se elimina por las heces. Los valores normales de uricosuria en adultos son 620 ± 75 mg/día1,2. Es preferible estudiar la eliminación de urato en forma de índice de excreción (normal: 0,40 ± 0,09 mg/100 ml)3 o, mejor, como excreción fraccional (normal: 7,25 ± 2,98%)4,5.
RECUERDO FISIOLÓGICO E HISTÓRICO DEL MANEJO TUBULAR RENAL DEL ÁCIDO ÚRICO. LOS NUEVOS AVANCES
A comienzos del siglo XX se suponía que el ácido úrico era filtrado por el riñón y, de este modo, se excretaba por la orina. Sin embargo, en 1950, Berliner et al. intentaron hallar una explicación al hecho, ya conocido, de que el aclaramiento de ácido úrico era menor que el de creatinina. Para dilucidar este «incomprensible fenómeno», dichos autores indujeron hiperuricemia, mediante una sobrecarga de carbonato de litio, a un grupo de sujetos sanos. Estudiando los aclaramientos de inulina y de urato, llegaron a la conclusión de que «el urato es excretado por filtración glomerular y por una reabsorción tubular activa» 6. Ese mismo año, Praetorius y Kirk describieron el caso de un paciente con una importante hipouricemia en el que el aclaramiento de ácido úrico era superior al de creatinina. Esto les hizo asumir que el riñón de dicho individuo secretaba ácido úrico. Fue el primer caso descrito de hipouricemia tubular renal7.
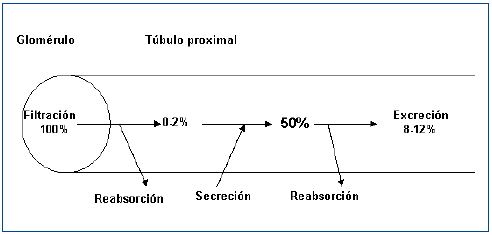
La existencia de una fase de secreción tubular de ácido úrico fue nuevamente propuesta en 1957 cuando Gutman y Yu estudiaron a 300 pacientes con gota y concluyeron que una reducción en la secreción tubular de ácido úrico explicaría la reducción de la uricosuria que mostraban8. Cuatro años más tarde, esos mismos autores publicaron su teoría de los tres componentes. El ácido úrico circulante en la sangre sería filtrado pasivamente en el glomérulo. Posteriormente, se reabsorbería activamente en el túbulo proximal y, a continuación, sería secretado hacia la luz tubular9. A principios de los años setenta, Diamond y Paolino, mediante la combinación secuencial de diversos uricosúricos (sulfinpirazona, probenecid y salicilatos a dosis elevadas) y pirazinamida, en sujetos sanos, señalaron la existencia de una reabsorción de urato que se localizaría en un lugar distal a la secreción (reabsorción postsecretora)10. La hipótesis de los tres componentes se había transformado en la de los cuatro componentes. El urato filtrado sería reabsorbido en el túbulo proximal en un 99-100% quedando en la luz tubular un 0-2% del urato filtrado. Posteriormente, se produciría una fase de secreción tubular, quedando en la luz tubular un 50% de la cantidad del urato inicialmente filtrado. Por último, se volvería a producir una reabsorción tubular proximal en una cifra cuantificada en el 80% del secretado. En fin, así se explicaría que la cantidad de ácido úrico excretada en la orina es, aproximadamente, el 10% de la cantidad de urato filtrado (figura 1). De este modo, los defectos en el manejo tubular del ácido úrico asociados con hipouricemia podían ser causados por defectos tanto de la reabsorción presecretora como de la reabsorción postsecretora aislados o combinados, o bien, por un incremento de la secreción tubular. Para establecer el mecanismo causal, se utilizaron pruebas farmacológicas con estímulo de pirazinamida (inhibiría la secreción tubular) y de probenecid o benzbromarona (inhibirían la reabsorción tubular postsecretora)11. Estas pruebas, actualmente, no se suelen realizar en la práctica diaria.
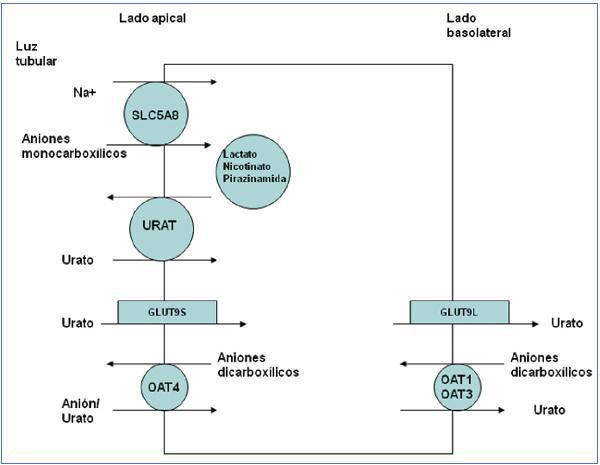
Los conocimientos sobre el metabolismo del acido úrico no cambiaron hasta que llegaron los nuevos avances consecuencia de la aplicación de las técnicas de biología molecular. Gracias a ellas, se han identificado varios transportadores y proteínas que han demostrado la complejidad del manejo del ión urato en el túbulo proximal12 (figura 2).
El transportador URAT1 que reabsorbe el urato filtrado fue identificado por Enomoto et al. en 200213. Está localizado en la membrana apical de las células del túbulo proximal y es codificado por el gen SLC22A12. URAT1 pertenece a la familia de transportadores de aniones orgánicos (OAT)14. En el riñón humano, el urato es transportado por medio del URAT1 a través de la membrana apical de las células tubulares proximales, en intercambio con aniones que son transportados hacia la luz tubular para mantener un balance eléctrico adecuado (figura 2). Se han descrito mutaciones en el gen SLC22A12 que codifica URAT1, en pacientes japoneses afectos de hipouricemia tubular renal15-17. También se han descritos mutaciones en ese gen en paciente coreanos18 y en tres familias israelitas de origen iraqui19. Estos pacientes se caracterizan por niveles muy bajos de ácido úrico con una excreción fraccional del mismo elevada (alrededor de 40-90%) y una respuesta atenuada de la uricosuria al probenecid y la pirazinamida15. Tanto losartán como benzbromarona ejercen su acción uricosúrica inhibiendo la acción de URAT120.
La salida del ácido úrico hacia el espacio peritubular se realiza mediante los transportadores basolaterales. En 2003, Jutabna et al. identificaron un nuevo transportador de iones orgánicos voltaje sensible, URATv1 (OATv1), que facilita la salida de urato de la célula21. Es codificado por el gen SLC2A922. Posteriormente, fue denominado como GLUT9 al conocerse que pertenece a una familia de proteínas facilitadoras del transporte de hexosas (fructosa, glucosa)23. Se han descrito dos variantes de la proteína: una isoforma GLUT9L que se expresa fundamentalmente en la membrana basolateral de las células del túbulo proximal y una isoforma GLUT9S que se expresa exclusivamente en la membrana apical de dichas células24, por lo que en los pacientes con esta otra variante de hipouricemia tubular renal la reducción en la reabsorción de urato ocurre en ambos lados de las células de los túbulos proximales renales25,26 (figura 2). En estos pacientes, la excreción fraccional de urato es superior a 150%26. Los portadores heterocigotos tienen niveles de urato moderadamente reducidos26.
GLUT9 es, seguramente, el principal regulador de los niveles de urato en humanos27. Así, se ha descrito que distintos polimorfismos en el gen SLC2A9 influyen en los niveles de urato en un amplio rango de valores28-30 por lo que, en el futuro, puede ser un objetivo terapéutico en pacientes con gota y con enfermedades cardiovasculares relacionadas25. Asimismo, se ha descrito la asociación entre ciertos polimorfismos del gen SLC2A9 y el desarrollo de nefrolitiasis31.
No obstante, la cuestión del manejo renal del ácido úrico es aún compleja. En 1985, Guggino y Aronson sugirieron que la pirazinamida estimulaba la reabsorción tubular de urato mediante un cotransporte Na+-dependiente32, es decir, lo contrario de lo conocido hasta ese momento, es decir, que inhibía la secreción tubular. Durante los años siguientes, se confirmó que existía un acoplamiento entre las reabsorciones de sodio y ácido en el túbulo proximal33. En 2004, se describió un candidato molecular para este tipo de cotransporte, SLC5A8, un transportador Na+-monocarboxilato que efectuaría un cotransporte Na+-dependiente de los aniones monocarboxílicos lactato, butirato, nicotinato, beta-hidroxibutirato y acetoacetato34,35. Curiosamente, se ha descrito que el gen humano SLC5A8 es un supresor tumoral. Su silenciamiento puede contribuir a la carcinogénesis y a la progresión de varios tumores. Pues bien, como aparece en la figura 2, SLC5A8 actuaría sinérgicamente con URAT1. Este mecanismo de reabsorción tubular apical de urato implicado con la reabsorción de Na+ explicaría la hiperuricemia inducida por los cetoácidos en la cetoacidosis diabética36, la intoxicación por etanol37, el tratamiento con pirazinamida38 o el síndrome metabólico39. Basta recordar que es conocido que la hiperinsulinemia incrementa la reabsorción de Na+40. Un incremento en las concentraciones séricas de estos aniones, una vez filtrados, aumentaría su reabsorción en el túbulo proximal que, a su vez, favorecería la reabsorción de urato al promover la actividad de URAT y el intercambio de esos aniones con el urato filtrado41.
En fin, la excreción por el riñón de una variedad de fármacos y metabolitos que son aniones orgánicos dicarboxilicos es mediada, mediante un intercambio con el urato, por una familia de transportadores multiespecíficos de aniones orgánicos OAT (genes OAT) que forman parte de la familia de transportadores de solutos SLC22. Diferentes OAT se localizan tanto en la membrana apical (OAT2, OAT4) como en la basolateral (OAT1 y OAT3) de los túbulos proximales renales42. Se ha sugerido que el probenecid tiene su efecto uricosúrico inhibiendo OAT412.
¿Y, mientras tanto, en qué ha quedado el modelo de los cuatro componentes? (figura 1). Su aceptación exige una separación anatómica de la reabsorción presecretora, la secreción tubular y la reabsorción postsecretora. En la actualidad, el mantenimiento de este esquema no se puede sostener, a falta de una detallada caracterización fisiológica y localización intrarrenal de los diferentes transportadores humanos enumerados anteriormente.
HIPOURICEMIA
La hipouricemia se diagnostica cuando los niveles plasmáticos de ácido úrico son menores o iguales a 2,0 mg/dl43, aunque Sperling propuso que debía utilizarse 2,1 mg/dl como limite bajo de la normalidad en mujeres y 2,5 mg/dl en hombres44. Se ha comunicado que se presenta en el 0,8% de los pacientes hospitalizados y en un 0,2% de la población general45.
En 1969 fue publicado un artículo realizado en el ámbito hospitalario en Toronto (Canadá), en el que se analizaba la utilidad clínica de 1.000 determinaciones de ácido úrico no solicitadas por los médicos responsables de los pacientes, pero que los bioquímicos incluían en los resultados analíticos46. Cuarenta y cuatro pacientes tenían niveles plasmáticos de ácido úrico menores de 2,6 mg/dl (un 4,4% de las muestras analizadas). En un solo caso el clínico se cuestionó la hipouricemia. En los 43 casos restantes, la hipouricemia se consideró irrelevante. Los diagnósticos de estos 44 pacientes fueron dispares y ninguno fue diagnosticado de causas clásicas de hipouricemia como el síndrome de Fanconi47 o la enfermedad de Wilson, por ejemplo.
El diagnóstico diferencial de la hipouricemia se realiza en función de la excreción fraccional de ácido úrico (tabla 1). La hipouricemia con una excreción fraccional de ácido úrico reducida se asocia con xantinuria, el tratamiento con alopurinol, las neoplasias y las alteraciones de la función hepática48. El tratamiento con rasburicasa también produce hipouricemia con excreción fraccional de ácido úrico reducida, dado que es un agente urolítico que cataliza la oxidación enzimática de ácido úrico a alantoína, un producto hidrosoluble, que se excreta fácilmente por vía renal.
Desde un punto de vista nefrológico, resulta de interés resaltar la hipouricemia asociada a xantinuria hereditaria (déficit autosómico recesivo de la enzima xantino-oxidasa), dado que es una hipouricemia severa menor a 1 mg/dl asociada a una disminución de la excreción fraccional de ácido úrico y a un aumento de la excreción de xantina y cuyo diagnóstico de confirmación se realiza mediante biopsia hepática o intestinal que demuestre disminución de la actividad enzimática.
La hipouricemia con una excreción fraccional de ácido úrico elevada está causada, principalmente, por hipouricemia de origen tubular renal, bien en forma de tubulopatía aislada15-19,25,26, o en seno de una tubulopatía compleja como el síndrome de de Toni-Debré-Fanconi causado por diversas entidades como la cistinosis, el síndrome de Lowe o la intoxicación por metales pesados. Otras causas son el uso de salicilatos, los contrastes intravenosos, la nutrición parenteral total, la enfermedad de Hodgkin y otras neoplasias, la enfermedad de Wilson y otras causas de cirrosis, la diabetes mellitus y el síndrome de secreción inadecuada de ADH. Por último, ha sido descrita su asociación con el hiperparatiroidismo49, la hiponatremia inducida por tiazidas50 y con la hiperbilirrubinemia51 (tabla 1). Es necesario recordar, también, que tanto los estrógenos, como el losartán, el dicumarol, los saliciltos a altas dosis y la trimetroprima-sulfametoxazol son fármacos que aumentan la excreción urinaria de ácido úrico.
Asimismo, desde un punto de vista nefrológico, se debe resaltar la existencia de hipouricemia en la diabetes mellitus, la hipouricemia asociada a la hiponatremia y la hipouricemia secundaria a tubulopatías.
Ha sido descrito que los pacientes con diabetes mellitus pueden tener hipouricemia56,57. Esa hipouricemia es observable tanto en la diabetes mellitus tipo 1, insulinodependiente58, como en la tipo 2, no insulinodependiente59,60, lo que implica que su fisiopatología debe estar relacionada, inicialmente, con alguna situación común a ambas entidades. La reducción en los niveles plasmáticos de ácido úrico se debe a un incremento de su aclaramiento renal58,61-63 y sólo se observa en pacientes con niveles normales de GFR. Cuando se realizaron pruebas farmacológicas de estímulo con uricosúricos, se describió tanto un defecto en la reabsorción presecretora68, una combinación del anterior con un defecto en la reabsorción postsecretora62,64 o este último defecto aislado62.
Incluso, en algunas comunicaciones, el aumento de la uricosuria se ha atribuido a una situación de hiperfiltración glomerular59,60, por lo que la hipouricemia podría ser un marcador de la aparición de nefropatía diabética60. Aunque no es constante, en general, se ha descrito una relación positiva entre la glucosuria y la uricosuria, de tal modo que existiría una interferencia entre la reabsorción tubular de glucosa y la capacidad tubular de reabsorción de urato61, por lo que la hipouricemia sería más probable en el caso de un mal control de la enfermedad. Así, la hipouricemia se asociaría con un mal control de la enfermedad, hiperfiltración o con un inicio tardío de la nefropatía69.
Respecto al valor de la hipouricemia en presencia de hiponatremia, la hipouricemia secundaria a expansión del volumen extracelular está asociada con una disminución de la reabsorción proximal de sodio y urato. Es frecuente en pacientes que reciben grandes cantidades de fluidos por vía intravenosa, que tienen polidipsia psicógena o en pacientes con SIADH. En estas situaciones, la restricción de agua corrige la hiponatremia y la hipouricemia. Sin embargo, la hipouricemia de los pacientes con enfermedad intracraneal y asociada con síndrome pierde-sal cerebral no se corrige con restricción de agua.
La hipouricemia no produce síntomas por sí misma. Los síntomas son los de la enfermedad causal. Sin embargo, la hipouricemia tubular aislada puede asociarse con nefrolitiasis y con fracaso renal agudo inducido por el ejercicio en pacientes portadores de mutaciones tanto en el gen URAT13,15-17 como en el GLUT925. En estos casos, la clínica de presentación es fatiga generalizada, náuseas o vómitos y malestar abdominal difuso; se suele presentar aproximadamente a las 2 semanas de haber realizado el ejercicio físico. Si el paciente ha sido previamente diagnosticado de hipouricemia su diagnóstico es sencillo y el tratamiento más temprano. Por ello, es recomendable que en las revisiones médicas rutinarias previas a la realización de ejercicio físico se determinen los niveles de ácido úrico, especialmente en asiáticos52. Se han propuesto dos mecanismos para explicar la producción del fallo renal agudo. En primer lugar, la nefropatía aguda por urato, causada por un incremento de su producción durante el ejercicio físico que culmina en su precipitación intratubular. El segundo mecanismo sería la agresión renal isquémica secundaria a la vasoconstricción de los vasos renales mediada por la producción de radicales libres de oxígeno durante el ejercicio53. Se sabe que el ácido úrico es el antioxidante soluble más abundante en humanos, de tal modo que se ha demostrado que preserva la función endotelial en las situaciones de estrés oxidativo54. Este mecanismo se apoya en los resultados histológicos que han demostrado necrosis tubular aguda55, aunque debe considerarse sólo como un cofactor desencadente, puesto que no se ha descrito fracaso renal agudo inducido por el ejercicio en pacientes con xantinuria26. Dinour et al. han propuesto un tercer mecanismo. Así, la acumulación de aniones no eliminados en los pacientes portadores de mutaciones tanto en el gen URAT como en el GLUT9 ejercería un efecto tóxico tubular que conduciría a una necrosis tubular aguda26.
Por último, queremos resaltar que es muy rara la asociación de hipouricemia tubular hereditaria e hipertensión arterial. dado que sólo se ha comunicado un caso en la bibliografía médica71.
CONCEPTOS CLAVE
1. Se define hipouricemia cuando los niveles plasmáticos de ácido úrico son menores o iguales a 2 mg/dl.
2. El diagnóstico diferencial de la hipouricemia se realiza habitualmente valorando la excreción fraccional de ácido úrico.
3. La hipouricemia con excreción fraccional de ácido úrico reducida se asocia con defectos en la producción de ácido úrico.
4. La hipouricemia con excreción fraccional de ácido úrico aumentada se asocia con defectos en el transporte tubular proximal de ácido úrico.
5. Actualmente se han producido avances en la identificación de los transportadores tubulares proximales de ácido úrico y en los genes que los codifican.
6. La hipouricemia es un marcador bioquímico de tubulopatía primaria o secundaria y de otras enfermedades subyacentes.

Figura 1. Representación esquemática de la hipótesis clásica de los cuatro componentes en el manejo tubular proximal del urato (% de urato filtrado).

Figura 2. El intercambiador URAT1 reabsorbe el urato filtrado en la membrana apical de las células del túbulo proximal en intercambio con aniones que son transportados hacia la luz tubular para mantener un balance eléctrico adecuado.

Tabla 1. Diagnóstico diferencial de hipouricemia según la excreción fraccional de ácido úrico



