





Durante la última década los progresos realizados en el conocimiento del genoma humano se han traducido en avances diagnósticos, pronósticos y terapéuticos en múltiples áreas de la medicina. El motivo de esta revisión es presentar, de una forma lo más actualizada y completa posible, los progresos obtenidos en el campo de las enfermedades autoinflamatorias sistémicas.
El concepto de enfermedades autoinflamatorias sistémicas fue propuesto en 1999 para definir una serie de entidades clínicas caracterizadas por procesos inflamatorios agudos, recurrentes o persistentes, en las cuales no se evidencian causas autoinmunitarias, infecciosas o neoplásicas1. A diferencia de las enfermedades autoinmunitarias, en las enfermedades autoinflamatorias sistémicas no se detectan autoanticuerpos a títulos elevados ni células T o B específicas de antígeno. En la actualidad se sabe que estas enfermedades son consecuencia de un proceso inflamatorio mal regulado y se consideran debidas a defectos en la respuesta inmunitaria innata2.
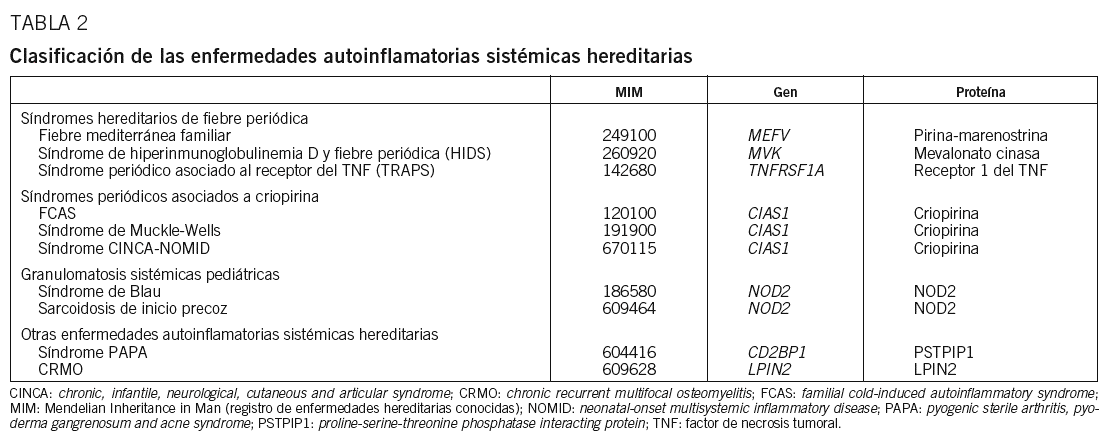
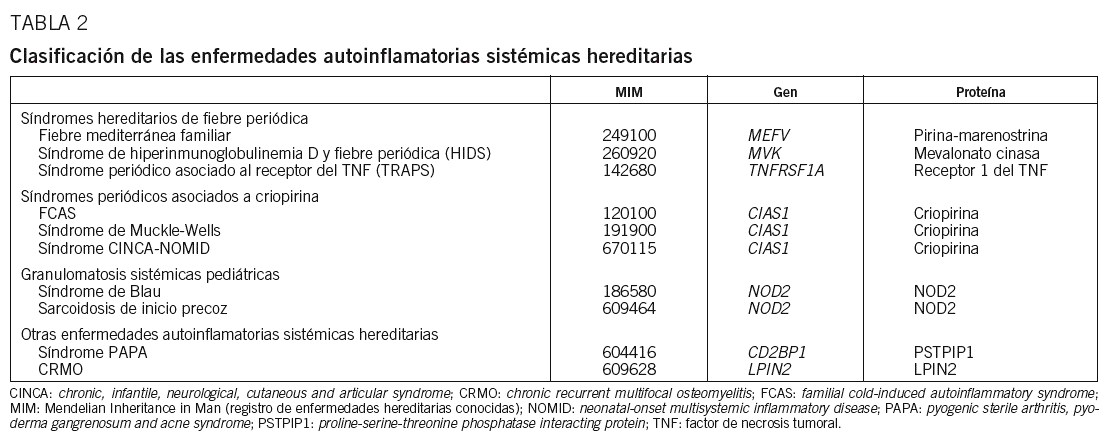
Desde la definición de enfermedad autoinflamatoria sistémica se han realizado varias propuestas con respecto a qué enfermedades debieran incluirse bajo este epígrafe (tabla 1)3,4. La presente revisión tiene por objetivo las formas hereditarias de las enfermedades autoinflamatorias sistémicas, en las que se ha descrito un patrón de herencia mendeliana e identificado el gen causante (tabla 2). Para ello esta revisión se ha dividido en 2 partes conceptualmente diferenciadas: en la primera se abordarán los síndromes hereditarios de fiebre periódica, mientras en la segunda se tratarán los síndromes periódicos asociados a criopirina, las granulomatosis sistémicas pediátricas hereditarias y el síndrome de artritis piogénica estéril, pioderma gangrenoso y acné (PAPA).

Por último, el lector debe tener presente que la clasificación aquí presentada no es definitiva, de tal manera que los avances en el conocimiento del proceso inflamatorio y de su regulación harán que determinadas enfermedades inflamatorias, consideradas en la actualidad enfermedades idiopáticas, puedan ser reevaluadas y reclasificadas a la luz de los nuevos hallazgos etiopatogénicos como enfermedades autoinflamatorias en un futuro más o menos cercano.
Síndromes hereditarios de fiebre periódica
Los síndromes hereditarios de fiebre periódica (HPFS) constituyen el subgrupo más importante dentro de las enfermedades autoinflamatorias sistémicas. Engloban 2 enfermedades con un patrón de herencia recesiva, la fiebre mediterránea familiar (FMF) y el síndrome de hiperinmunoglobulinemia D y fiebre periódica (HIDS), y una entidad con herencia dominante, el síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAPS)4. Todas estas entidades deben considerarse enfermedades raras, dada su baja incidencia (< 5 casos por cada 10.000 habitantes), si bien la incidencia mundial de la FMF es muy superior a la del resto de HPFS. No obstante, debemos dejar constancia de que entre la población española se ha identificado a pacientes afectados de todos y cada uno de estos HPFS.
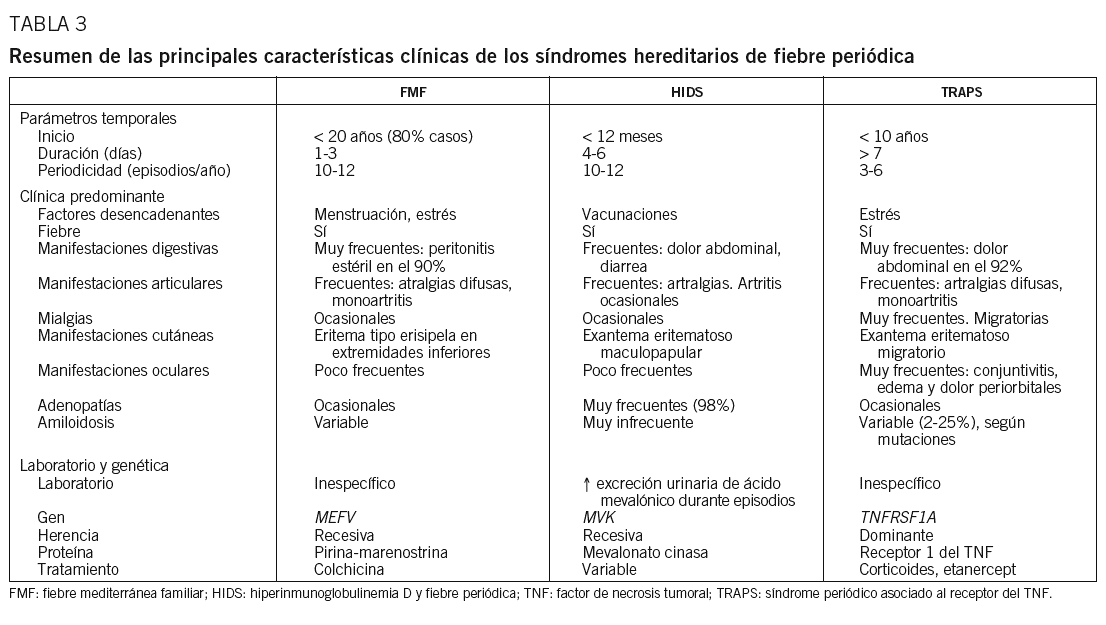
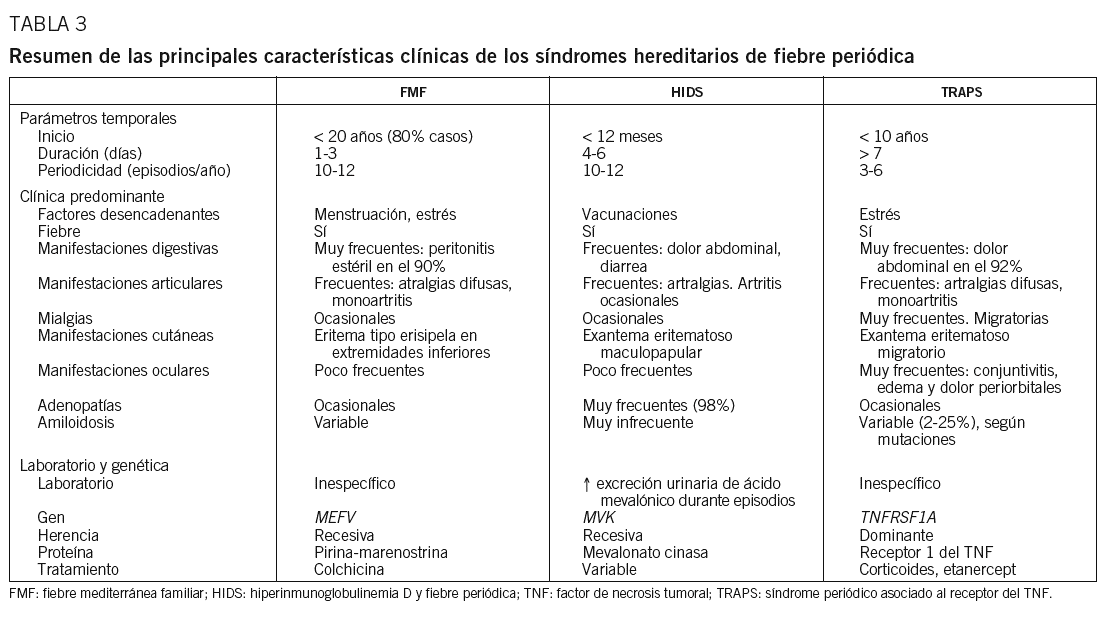
La característica común a estas 3 enfermedades es la aparición de episodios inflamatorios agudos, autolimitados y de duración variable, que recurren a lo largo del tiempo, con una periodicidad diferente para cada una de ellas, de tal modo que pueden identificarse unos parámetros temporales (edad de inicio, duración de los episodios agudos y periodicidad), que serán de gran utilidad en el diagnóstico diferencial de estas entidades (tabla 3). Durante los períodos intercrisis los pacientes no presentan síntomas clínicos y, por otro lado, es frecuente la detección de cierto grado de inflamación subclínica, que se refleja en alteraciones de los parámetros analíticos.
Fiebre mediterránea familiar
La FMF (MIM 249100; MIM es el acrónimo de Mendelian Inheritance in Man, registro donde se incluyen todas las enfermedades hereditarias conocidas, y el número que aparece a continuación es el número de referencia con que la enfermedad ha sido introducida en dicho registro, que puede consultarse en: http://www.ncbi.nlm.nih.gov/) es la enfermedad autoinflamatoria sistémica más frecuente en todo el mundo y representa el prototipo de HPFS. Las primeras descripciones clínicas datan de comienzos del siglo xx, si bien bajo nombres tan dispares como poliserositis recurrente, peritonitis periódica y poliserositis recurrente hereditaria. El nombre de FMF se propone a mediados del siglo xx y recoge 3 características de la enfermedad que se consideran clave: a) clínica de fiebre recurrente; b) alta incidencia de la enfermedad en poblaciones de la cuenca mediterránea, y c) presencia de historia familiar, con un patrón de herencia autosómica recesiva5,6.
Desde el punto de vista demográfico, en la actualidad se han dado a conocer casos de FMF tanto en poblaciones mediterráneas como en poblaciones no mediterráneas7-10. Asimismo, en la cuenca mediterránea puede observarse un gradiente este-oeste de incidencia de la enfermedad, de tal manera que determinadas poblaciones del Mediterráneo oriental, denominadas «poblaciones ancestrales» (judíos, árabes, armenios y turcos), presentan una elevada incidencia de la enfermedad, mientras que ésta disminuye en regiones del Mediterráneo central y occidental (griegos, italianos, franceses y españoles). Así, en la población española la FMF es también el HPFS más frecuente y afecta a individuos sin un origen étnico específico. En este sentido debemos señalar que la pertenencia a una de las poblaciones consideradas ancestrales debe reforzar la sospecha diagnóstica de la enfermedad, mientras que la falta de pertenencia no debe considerarse un factor excluyente para su diagnóstico.
Los pacientes afectados de FMF presentan episodios inflamatorios breves (12-72 h, ligeramente más prolongados si hay afectación articular), que tienden a recurrir cada 4-5 semanas. Habitualmente la enfermedad comienza durante la infancia o adolescencia, de tal manera que en el 80% de los pacientes los primeros episodios inflamatorios suelen observarse antes de los 20 años de edad4,5,11. Se han identificado algunos factores desencadenantes de los episodios, tales como la menstruación, la ovulación, el puerperio y situaciones de estrés psicológico o actividad física intensa, así como determinados factores protectores, como el embarazo. En este sentido, parece lógico afirmar que el componente hormonal puede desempeñar un papel modulador en la aparición de manifestaciones clínicas de la enfermedad en las mujeres. Asimismo, se han identificado determinados síntomas prodrómicos de los episodios, tales como escalofríos, malestar general y molestias abdominales.
Desde el punto de vista clínico, los episodios inflamatorios de los pacientes afectados de FMF se caracterizan por fiebre recurrente asociada a poliserositis y sinovitis inflamatoria, así como a determinadas lesiones cutáneas4-6,11,12. Las manifestaciones pueden variar de un episodio a otro en el mismo individuo y también pueden ser diferentes entre individuos distintos, aun cuando pertenezcan a una misma familia.
La fiebre suele presentarse como una elevación súbita de la temperatura corporal, que puede alcanzar los 40-41 ºC, y una disminución ligera, y está presente a lo largo de todo el episodio inflamatorio. Las serositis observables son típicamente inflamatorias, con un importante infiltrado de polimorfonucleares, y las serosas afectadas con mayor frecuencia son el peritoneo y la pleura, mientras que la afectación pericárdica y meníngea es más infrecuente5,6.
La peritonitis inflamatoria está presente en la práctica totalidad de los pacientes, al menos en alguno de sus episodios inflamatorios. Clínicamente se traduce por dolor abdominal, que puede ser difuso o localizado en un cuadrante, de intensidad variable, desde leves molestias hasta un cuadro de abdomen agudo, con rigidez de la pared muscular y signo de rebote positivo5,6. En los pacientes que han precisado cirugía mayor como consecuencia del dolor abdominal se ha observado una laparotomía blanca, con peritonitis inflamatoria estéril. La peritonitis suele provocar asimismo un enlentecimiento del peristaltismo intestinal, de tal manera que es más frecuente la aparición de estreñimiento que de diarrea.
La afectación pleural, que puede estar presente hasta en el 50% de los pacientes, se traduce habitualmente en dolor torácico, y en las radiografías de tórax pueden visualizarse pequeños derrames pleurales o atelectasias5,6.
La sinovitis inflamatoria está presente en un porcentaje variable de los pacientes (50-75%). La manifestación articular más frecuente son las artralgias, generalmente difusas. La artritis se observa en un porcentaje menor de casos, habitualmente es monoarticular y afecta preferentemente a grandes articulaciones, como el tobillo, la rodilla y la cadera13. El líquido sinovial en dichas artritis suele ser estéril, con abundantes polimorfonucleares, y las pruebas de imagen ponen de manifiesto la ausencia de cambios óseos. Cuando el componente artrítico está presente, la duración de los episodios inflamatorios tiende a ser mayor, si bien en estos casos la fiebre y el resto de síntomas suelen desaparecer a las 72 h de su inicio.
La lesión cutánea más característica de los pacientes con FMF es el eritema de tipo erisipeloide, habitualmente localizado en la parte anterior de la pierna o en el dorso del pie, de manera unilateral o bilateral. No obstante, su frecuencia es inferior al resto de manifestaciones antes comentadas, que oscila entre el 3 y el 45% de los pacientes4-6.
Además de las manifestaciones anteriores, en los pacientes afectados de FMF se ha descrito una serie de manifestaciones infrecuentes, que deben tenerse en cuenta. Entre ellas debemos destacar: a) inflamación escrotal aguda, habitualmente unilateral, que suele aparecer en niños en edad prepuberal y que es consecuencia de una inflamación de la túnica vaginalis; b) mialgias; c) cefalea, secundaria a una irritación meníngea de causa inflamatoria, que ocasionalmente puede generar convulsiones y cambios en el electroencefalograma; d) pericarditis inflamatoria, y e) determinadas formas de vasculitis (púrpura de Henoch-Schonlein, poliarteritis nodosa)5,6,14-17.
En los intervalos intercrisis los pacientes no presentan síntomas febriles ni inflamatorios, si bien algunos pueden referir febrícula o ciertas molestias abdominales. Sin embargo, en estos intervalos asintomáticos pueden observarse valores elevados de los parámetros bioquímicos que señalan inflamación, especialmente la velocidad de sedimentación globular (VSG), la proteína C reactiva y la proteína sérica del amiloide (SAA1), así como cambios moderados en el hemograma. Todos estos cambios denotan una situación de inflamación subclínica como consecuencia del defecto que genera la enfermedad5,18,19.
La principal y más temida complicación de la FMF es la amiloidosis secundaria, que afecta a un porcentaje bajo de pacientes (< 5%), si bien su incidencia es menor desde la instauración de la colchicina como tratamiento de los episodios inflamatorios de la FMF. La amiloidosis secundaria se debe al depósito de amiloide en los tejidos y órganos de la economía, y habitualmente se presenta por encima de la tercera-cuarta décadas de la vida5,20,21. La sustancia amiloide depositada procede de la degradación de la proteína SAA1, un reactante de fase aguda de síntesis hepática, y su depósito es consecuencia de procesos inflamatorios repetidos y no controlados durante años20,21. La forma clínica de presentación más frecuente es la afectación renal, en forma de síndrome nefrótico20,21. Otras localizaciones frecuentes del depósito en las amiloidosis secundarias son el intestino, el hígado, el bazo, el tiroides y las glándulas suprarrenales20,21. A diferencia de otras formas de amiloidosis, son muy raras las neuropatías o las cardiopatías. Entre los factores de riesgo para el desarrollo de amiloidosis secundaria en los pacientes afectados de FMF se encuentran: a) sexo masculino; b) homocigosidad para la mutación M694V; c) ser portador del genotipo */* en el gen que codifica la proteína SAA1, y d) presentar episodios con importante componente artrítico22.
Asimismo debemos señalar que en un porcentaje muy pequeño de pacientes la FMF, denominada del tipo II, comienza de forma tardía con síntomas derivados de la amiloidosis secundaria, sin que previamente se hayan presentado los episodios inflamatorios recurrentes11.
Al igual que en el resto de las enfermedades autoinflamatorias sistémicas hereditarias, no hay un marcador hematológico o bioquímico específico o patognomónico de la FMF. Durante los episodios inflamatorios agudos es habitual identificar leucocitosis secundaria a neutrofilia, habitualmente con desviación izquierda, trombocitosis y cierto grado de anemia de procesos crónicos. En la esfera bioquímica, se observa una intensa reacción de fase aguda durante los episodios, caracterizados por la elevación de la VSG y de los valores plasmáticos de la proteína C reactiva, haptoglobina, fibrinógeno, ferritina y proteína SAA111. Durante los intervalos clínicamente asintomáticos pueden detectarse tanto normalización como elevaciones moderadas de estos parámetros18,19.
Genética. Tras las primeras descripciones de la enfermedad quedó de manifiesto el componente hereditario de la FMF y se señaló un patrón hereditario autosómico recesivo. En 1997 varios grupos, agrupados en los denominados Consorcio Internacional y Consorcio Francés, establecieron las bases genéticas causantes de la FMF. Ambos grupos identificaron mutaciones responsables de la enfermedad en un gen anteriormente no conocido, que recibió el nombre de MEFV (por MEditerranean FeVer) y que codifica para una proteína nueva, de 781 aminoácidos, a la que el Consorcio Internacional denominó pirina (del griego pyros, «fiebre») y el Consorcio Francés, marenostrina (del latín mare nostrum, «mar Mediterráneo»)23,24. Desde entonces se han identificado más de 70 mutaciones asociadas a enfermedad en dicho gen, que se encuentran disponibles en la base de datos INFEVERS (dirección electrónica: http://fmf.igh.cnrs.fr/infevers)25 y que pueden dividirse en 2 grandes grupos:
1. Mutaciones recurrentes: mutaciones que aparecen en distintos individuos, pertenecientes a poblaciones diferentes, se originaron hace cientos de años y se diseminaron desde su lugar de aparición con los movimientos migratorios humanos. Se postula que la pervivencia de dichas mutaciones a lo largo del tiempo ha podido tener un efecto positivo en la selección natural, al conferir algún efecto protector frente a algún proceso infeccioso, hoy día desconocido18,23. En este apartado podríamos diferenciar a su vez 2 grupos:
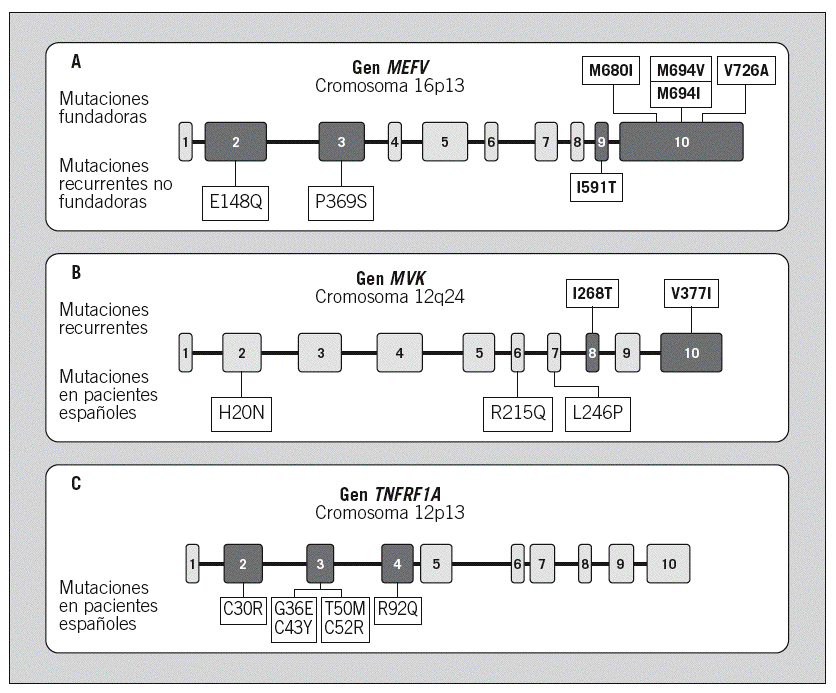
Mutaciones fundadoras, que se postula aparecieron en Babilonia hace unos 2.000 años y que son las mutaciones más frecuentemente identificadas en cualquier población mundial, incluidas las que hemos detectado en pacientes de la población española. Estas mutaciones fundadoras son la Met-680-Ile (M680I), Met-694-Val (M694V), Met-694-Ile (M694I) y Val-726-Ala (V726A), todas ellas localizadas en el exón 10 del gen MEFV (fig. 1A)7-10,23,24,26-28.
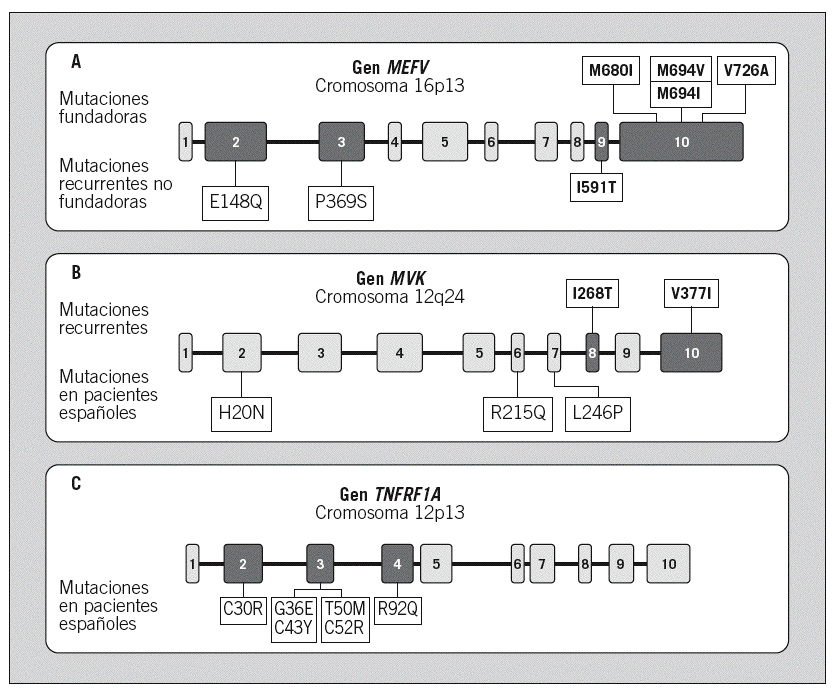
Fig. 1. Organización genómica de los genes asociados a los diferentes síndromes hereditarios de fiebre recurrente. En negro se señalan los exones donde se concentran las mutaciones asociadas a enfermedad en cada uno de los genes. A: gen MEFV, asociado a la fiebre mediterránea familiar. En la parte superior se detalla la localización de las mutaciones recurrentes fundadoras, y en la parte inferior, la localización de las mutaciones recurrentes no fundadoras. B: gen MVK, asociado al síndrome de hiperinmunoglobulinemia D y fiebre periódica (HIDS). En la parte superior se localizan las mutaciones recurrentes identificadas en el HIDS, y en la parte inferior, las mutaciones privadas identificadas por nuestro grupo en pacientes españoles afectados de HIDS. C: gen TNFRSF1A, asociado al síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAPS). En la parte inferior se señala la localización de las mutaciones identificadas por nuestro grupo en pacientes españoles afectados de TRAPS.
Mutaciones recurrentes no fundadoras, que aparecen en múltiples poblaciones, sin un origen geográfico y temporal tan bien definido como en las anteriores. En este grupo se encontrarían las siguientes mutaciones: Glu-148-Gln (E148Q), el alelo complejo P369S-R408Q, Ile-591-Thr (I591T), Lys-695-Arg (K695R), Ala-744-Ser (A744S) y Arg-761-His (R761H)7-9,26-30.
2. Mutaciones no recurrentes: mutaciones que aparecen en un individuo o en una determinada familia de forma aislada, de origen reciente y sin repercusión en el conjunto de la población. También se denominan «mutaciones privadas». Algunos ejemplos de ellas serían las siguientes mutaciones, identificadas todas ellas por nuestro grupo en pacientes españoles: Glu-163-Ala (E163A), Pro-180-Arg (P180R), Ala-268-Val (A268V), Glu-319-Lys (E319K), His-478-Tyr (H478Y) y Ile-640-Met (I640M)9.
El patrón de herencia descrito clásicamente para la FMF es autosómico recesivo y, en consecuencia, requeriría que tanto la copia de origen paterno del gen como la materna presenten mutaciones. Sin embargo, los estudios genéticos llevados a cabo en pacientes con diagnóstico clínico de FMF ponen de manifiesto que un porcentaje importante de ellos (hasta un 40%) son portadores de una única mutación en el gen MEFV, mientras que otros no portan ninguna27-30. Asimismo, se han descrito unas pocas familias portadoras de mutaciones que se comportan claramente con un patrón de herencia dominante31,32. Esta heterogeneidad genética observable en la FMF puede deberse a la existencia de genes, en la actualidad desconocidos, que, cuando están mutados, generan un fenotipo clínico similar al observado en la FMF.
Etiopatogenia. En la actualidad se desconoce con precisión la función o funciones llevadas a cabo por la proteína pirina-marenostrina (P/M), codificada por el gen MEFV. Si se analiza la expresión del gen en el sistema hematopoyético (expresión elevada en neutrófilos, monocitos, eosinófilos, células dendríticas, pero no en linfocitos), puede proponerse un importante papel en la regulación de la respuesta inmunitaria innata33,34. Asimismo, los estudios de la estructura de la proteína P/M han identificado un dominio estructural nuevo, denominado dominio pirina (PyD). Dicho PyD presenta semejanza con los miembros de la familia de dominios de muerte celular, implicados todos ellos en los procesos de regulación de la apoptosis35.
En la actualidad se proponen las siguientes hipótesis sobre la función de la P/M, que no deben contemplarse como excluyentes: a) hipótesis de secuestro, en la que la P/M actuaría como un regulador negativo del inflamasoma, dado que la P/M, a través de su PyD, podría interactuar con la proteína ASC (apoptosis-associated speck-like protein containing a CARD), con lo que impediría su acoplamiento con las demás proteínas que constituyen el inflamasoma y provocaría una disminución de la producción de la forma activa de la caspasa-1, y b) hipótesis del inflamasoma de P/M, en la que una de las regiones de la P/M, denominada dominio B30.2, podría actuar como receptor de patrones moleculares asociados a patógenos y, tras su unión, podrían producirse cambios conformacionales en la proteína que podrían dar lugar a la interacción directa de P/M con la procaspasa 1, generándose la forma activa de la caspasa 1 y, en consecuencia, el procesamiento de las procitocinas inflamatorias36,37.
Tratamiento. En 1972-1974 quedó establecida la eficacia de la colchicina por vía oral para impedir la reaparición de los episodios inflamatorios o, en su defecto, para disminuir la intensidad de los síntomas y su frecuencia, así como su papel protector en el desarrollo de amiloidosis secundaria38-42. En aquellos trabajos quedó establecido que dicho tratamiento debe ser diario, continuado, con dosis en adultos que oscilan entre 1 y 2 mg/día, si bien algunos pacientes pueden presentar efectos beneficiosos a dosis inferiores y otros requerir dosis superiores para alcanzarlos. La administración intermitente de la colchicina puede tener cierto efecto beneficioso en lo referente a los episodios inflamatorios, pero no así en el papel protector del desarrollo de amiloidosis secundaria. Asimismo, la administración puntual de la colchicina durante un episodio agudo no suele tener ningún efecto. En cuanto a los efectos secundarios del tratamiento, el más frecuente es la aparición de intolerancia digestiva, en forma de diarrea, posiblemente debida a la aparición de intolerancia a la lactosa. Para evitar o disminuir este efecto secundario se recomienda la introducción gradual y ascendente del fármaco, asociado con una dieta baja en lactosa. Se han dado a conocer otros efectos secundarios menos frecuentes del tratamiento con colchicina, tales como miopatías, neuropatías e incremento de los valores plasmáticos de creatinina5,43,44.
Un porcentaje pequeño (en torno al 5%) de los pacientes con FMF adecuadamente tratados con colchicina oral pueden no responder a dicho tratamiento. En estos casos se han propuesto alternativas terapéuticas, tales como la administración de interferón alfa, colchicina por vía intravenosa a dosis bajas y agentes biológicos bloqueadores del factor de necrosis tumoral (TNF) o de la interleucina 1ß11.
Síndrome de hiperinmunoglobulinemia D y fiebre periódica
El HIDS (MIM 260920) es un síndrome de fiebre recurrente, con un patrón de herencia recesiva, que fue descrito como una entidad clínica independiente en 1984 por un grupo holandés, razón por la cual también se conoce como «fiebre holandesa»45. Sin embargo, desde el punto de vista epidemiológico, se han descrito casos de pacientes con HIDS en poblaciones diferentes de la holandesa, incluida la española, motivo por el cual, a semejanza de lo comentado para la FMF, el factor étnico no debe considerarse un factor excluyente para el diagnóstico46-49.
Los pacientes de HIDS presentan unos 10-12 episodios inflamatorios anuales, que duran 4-5 días y característicamente comienzan muy temprano en la vida, de tal forma que en la mayoría de los pacientes puede identificarse el primer episodio antes de los 12 meses de edad4,11,12. Las vacunaciones aparecen como uno de los principales factores desencadenantes de los episodios4,50,51. Asimismo, se ha constatado que los episodios inflamatorios tienden a ser menos intensos y menos frecuentes en el tiempo cuando los pacientes alcanzan la edad adulta11,50,51. Ésta podría ser una explicación de la práctica ausencia de amiloidosis secundaria en este grupo de afectados de HPFS.
Desde el punto de vista clínico, los episodios inflamatorios se caracterizan por fiebre recurrente, acompañada de adenopatías y manifestaciones cutáneas, digestivas y articulares principalmente. A semejanza del patrón febril observado en la FMF, el patrón de la fiebre recurrente observado en el HIDS presenta un incremento rápido de la temperatura corporal, que puede alcanzar los 40-41 ºC, para después mantenerse a lo largo del episodio inflamatorio y disminuir de forma lenta. En muchos de los pacientes los escalofríos pueden preceder o acompañar al incremento de temperatura.
Las adenopatías están presentes hasta en el 98% de los pacientes con HIDS. Son fácilmente palpables en la región laterocervical, unilaterales o bilaterales, dolorosas a la palpación y en muchos de los pacientes hacen preciso un diagnóstico diferencial con un proceso proliferativo. Los estudios anatomopatológicos de dichas adenopatías demuestran que son de naturaleza inflamatoria50,51.
Las manifestaciones cutáneas están presentes hasta en el 85% de los pacientes; el exantema eritematoso y maculopapular, con grados variables de afectación de la superficie corporal, es la forma más frecuentemente detectada50,51.
Entre las manifestaciones digestivas destaca la aparición de náuseas y vómitos (88%), dolor abdominal (83%) y diarrea (68%). A semejanza de lo descrito para la FMF, el dolor abdominal es consecuencia de una peritonitis inflamatoria y puede ser tanto difuso como localizado en algún cuadrante, de intensidad variable, pero por lo general inferior a la intensidad del dolor abdominal de los episodios inflamatorios observados en la FMF4,11,12,50,51. Posiblemente ésta es una de las razones por las cuales el número de pacientes que precisan cirugía mayor es inferior en el HIDS que en la FMF.
Entre las manifestaciones osteomusculares destacan la presencia de artralgias difusa (88%) y de artritis (71%), habitualmente monoarticular.
Se han dado a conocer diversas manifestaciones menos frecuentes que pueden acompañar a las anteriores, tales como aftas orales, dolor faríngeo, esplenomegalia, cefalea y mialgias generalizadas50,51. Por otro lado, debemos destacar que los pacientes con HIDS no suelen presentar afectación neurológica ni anomalías morfológicas50,51. A diferencia de lo que ocurre en el resto de HPFS, se han comunicado muy pocos casos de amiloidosis secundaria entre pacientes afectados de HIDS, posiblemente como consecuencia de la atenuación de los episodios inflamatorios durante la edad adulta52,53.
Entre los parámetros hematológicos y bioquímicos asociados a los episodios inflamatorios, debemos diferenciar entre aquellos no específicos del HIDS, compartidos por otros HPFS (la leucocitosis secundaria a neutrofilia, la trombocitosis y el aumento de los valores plasme pueden resumirse en:
Aumento policlonal de inmunoglobulina D superior a 100 U/ ml (> 14 mg/dl) en 2 determinaciones separadas por un intervalo mínimo de un mes. Dicho aumento policlonal de inmunoglobulina D generalmente se acompaña de un aumento policlonal de inmunoglobulina A. Esta alteración analítica está presente en la gran mayoría de los pacientes afectados de HIDS, pero debemos señalar que no es ni específica ni patognomónica de éste, por cuanto aumentos por encima del límite de normalidad pueden observarse en múltiples enfermedades inflamatorias, de tal manera que en la actualidad se considera un epifenómeno del proceso inflamatorio.
Aumento de la excreción urinaria de ácido mevalónico durante los episodios inflamatorios agudos, no así durante los intervalos asintomáticos. Este parámetro analítico se considera el único parámetro de laboratorio que permite diferenciar el HIDS del resto de HPFS4,11,50,51,54.
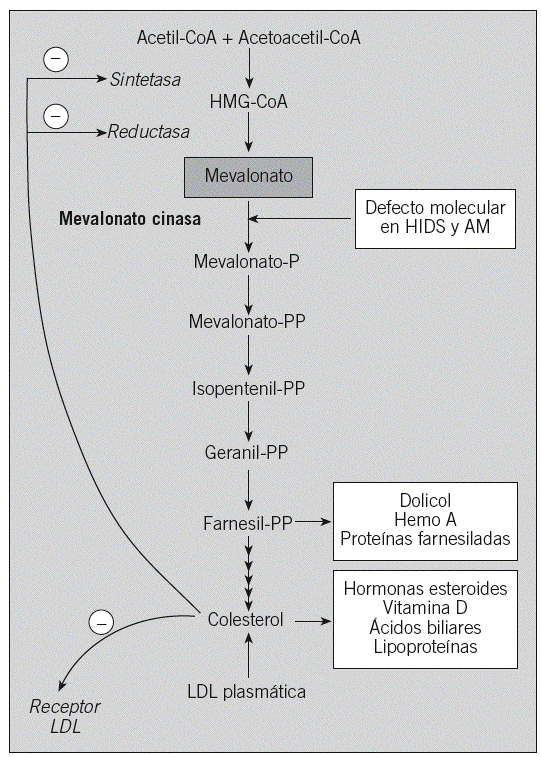
Genética. Las bases genéticas responsables del HIDS fueron identificadas en 1999 por 2 grupos independientes, al demostrar la asociación entre mutaciones en el gen mevalonato cinasa (MVK) y la enfermedad54,55. El gen MVK codifica para la mevalonato cinasa, una enzima clave en la síntesis de colesterol y de compuestos isoprenilados (fig. 2). Con anterioridad se había demostrado que el gen MVK era responsable de una enfermedad metabólica, la aciduria mevalónica, un trastorno grave que presenta alteraciones tanto neurológicas como morfológicas y muchos de los signos y síntomas observados en el HIDS, tales como fiebre recurrente, adenopatías y manifestaciones cutáneas y digestivas56. En los últimos años se han descrito unos pocos casos en que el HIDS y la aciduria mevalónica se solapan, de tal manera que se ha propuesto que ambas entidades clínicas corresponderían a las formas extremas de un espectro de gravedad continuo, representando la aciduria mevalónica la forma más grave y el HIDS, la más leve57.
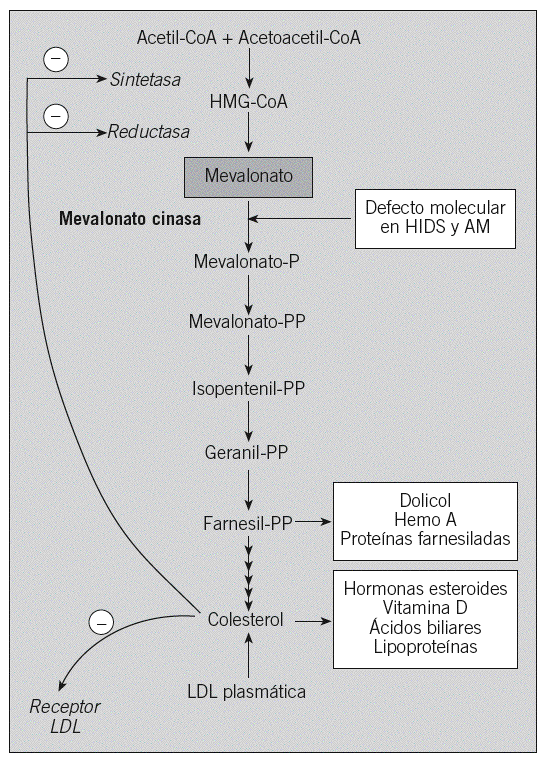
Fig. 2. Ruta metabólica del colesterol y de los compuestos isoprenilados. Se señala la enzima mevalonato cinasa, cuyos defectos están asociados al síndrome de hiperinmunoglobulinemia D y fiebre periódica (HIDS) y a la aciduria mevalónica (AM). CoA: coenzima A; HMG: hidroximetilglutaril; LDL: lipoproteína de baja densidad.
Desde el punto de vista genético, en el HIDS están sobrerrepresentadas 2 mutaciones, denominadas Ile-268-Thr (I268T) y Val-377-Ile (V377I), de tal manera que la mayoría de los pacientes con HIDS son portadores de una o ambas, mientras que dichas mutaciones están ausentes en la aciduria mevalónica (fig. 1B)49,54,55,58. Una posible explicación de este hecho vendría dada por la repercusión de la mutación en la funcionalidad de la proteína mevalonato cinasa. En este sentido, se ha demostrado que las mutaciones del gen MVK asociadas a aciduria mevalónica generan una enzima funcionalmente inactiva, mientras que las mutaciones asociadas a HIDS generan una enzima con una actividad enzimática baja (5-10%), pero no ausente58.
Etiopatogenia. En lo referente al mecanismo etiopatogénico subyacente en el HIDS, se desconoce en detalle cómo un defecto en la ruta metabólica del colesterol y de compuestos isoprenilados puede dar lugar a un síndrome de fiebre recurrente. Varios trabajos han puesto de manifiesto, por un lado, la disminución importante de los compuestos isoprenilados en los pacientes con HIDS y, por otro, el efecto regulador negativo de dichos compuestos, y en especial del geranilgeraniol, sobre el procesamiento de la procaspasa-1 en el inflamasoma59,60. Así pues, una de las hipótesis propondría que la disminución de la formación de los compuestos isoprenilados en los pacientes afectados de HIDS daría lugar a la pérdida del efecto regulador negativo de aquéllos sobre el inflamasoma, con lo que se generaría una mayor cantidad de caspasa-1 activa y de citocinas proinflamatorias, con las consecuencias biológicas que ello conlleva.
Tratamiento. No existe un tratamiento de elección para estos pacientes. Se han comunicado respuestas clínicas positivas con una gran cantidad de fármacos, como la talidomida, los antiinflamatorios no esteroideos, los glucocorticoides, las estatinas, los agentes biológicos bloqueadores del TNF o de la interleucina 1ß4,61-66.
Síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAPS)
El acrónimo TRAPS (del inglés TNF receptor-associated periodic syndrome; MIM 142680) fue propuesto en 1999 para definir un síndrome de fiebre periódica, de herencia dominante, que es debido a mutaciones en el receptor 1 del TNF1. Dicho síndrome se había descrito desde el punto de vista clínico cierto tiempo atrás y recibió diferentes denominaciones en función del grupo que lo describió, tales como fiebre hiberniana familiar (para denotar el grupo étnico donde se describió, en contraposición a la FMF) y síndrome de fiebre periódica autosómica dominante con amiloidosis, denominación que refleja una de las complicaciones más frecuentes de dicho síndrome67-69.
Desde el punto de vista epidemiológico, si bien los primeros casos se describieron en población irlandesa y centroeuropea, se han comunicado también casos aislados y casos familiares en múltiples poblaciones de todo el mundo, de orígenes étnicos muy diversos, incluida la población española, de tal modo que, a semejanza de lo comentado para los otros HPFS, el factor étnico no debe considerarse un factor decisivo en el diagnóstico de esta enfermedad69-73.
Los pacientes con TRAPS presentan episodios inflamatorios agudos, de duración prolongada (> 7 días, y puede alcanzar las 3-4 semanas de duración), que comienzan habitualmente antes de los 10 años de edad y tienen una periodicidad más variable que en los otros HPFS, de modo que algunos afectados llegan a presentar entre 4 y 8 episodios anuales3,4,11,12,74. No obstante, algunos pacientes pueden referir síntomas de forma persistente, con exacerbaciones y atenuaciones en su intensidad, sin claros intervalos asintomáticos. La distribución por sexos tiende a ser 1:1, como cabría esperar de una enfermedad con un patrón de herencia autosómica dominante, y, a semejanza de lo expuesto para el resto de HPFS, algunos pacientes pueden referir determinados factores o situaciones como desencadenantes de los episodios (estrés físico, psicológico, ovulación y menstruación), y en algunos casos refieren determinados síntomas como prodrómicos de las crisis (edema periorbital, malestar general, cefalea)74.
Desde el punto de vista clínico se puede diferenciar una serie de síntomas frecuentes, presentes en la mayoría de los episodios inflamatorios del TRAPS (fiebre recurrente, manifestaciones osteomusculares, cutáneas, oculares y digestivas), y otros menos frecuentes.
La fiebre recurrente se observa en la gran mayoría de los pacientes con TRAPS y suele alcanzar los 40-41 ºC. Si bien no se ha observado un patrón febril típico, en ocasiones puede identificarse un perfil en picos y valles a lo largo de todo el episodio, siendo habituales las elevaciones vespertinas de la temperatura corporal. A modo de curiosidad, debemos mencionar que recientemente se han descrito algunos casos de pacientes con TRAPS que no presentan fiebre recurrente entre los signos clínicos objetivables durante los episodios agudos75.
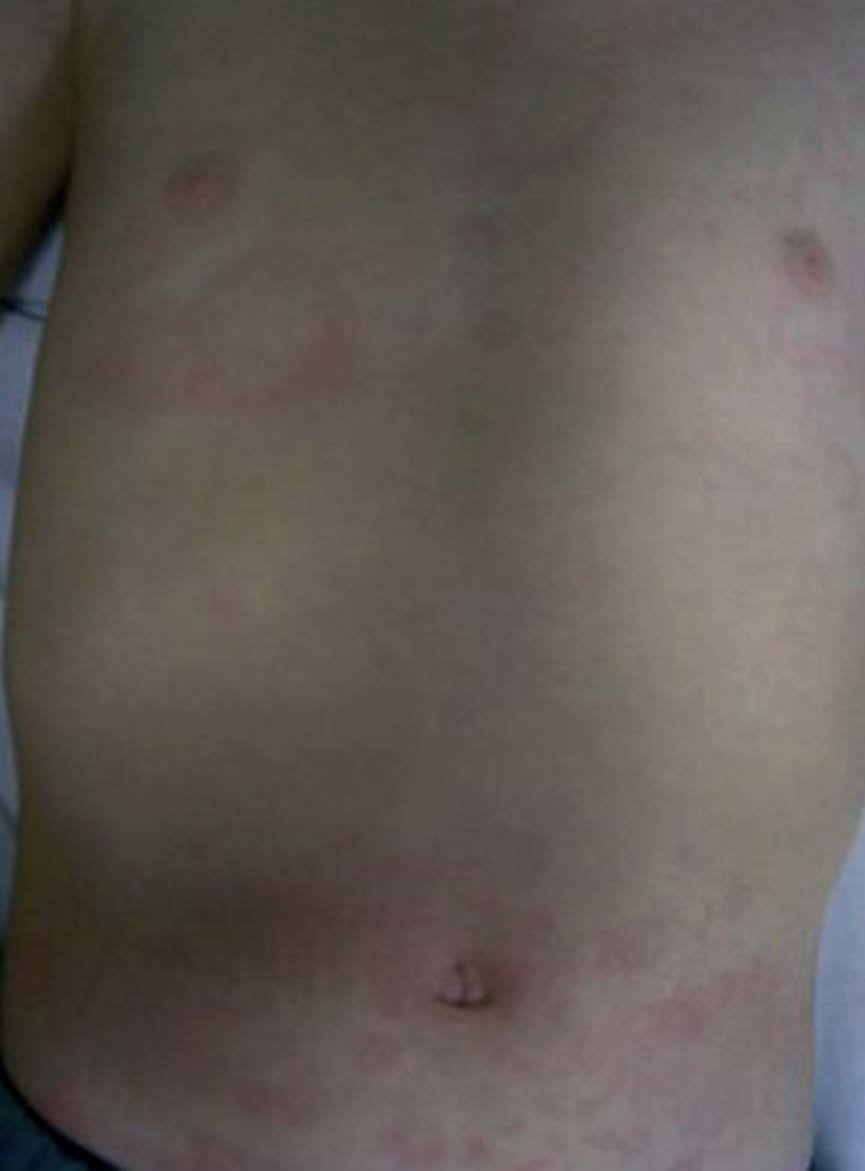
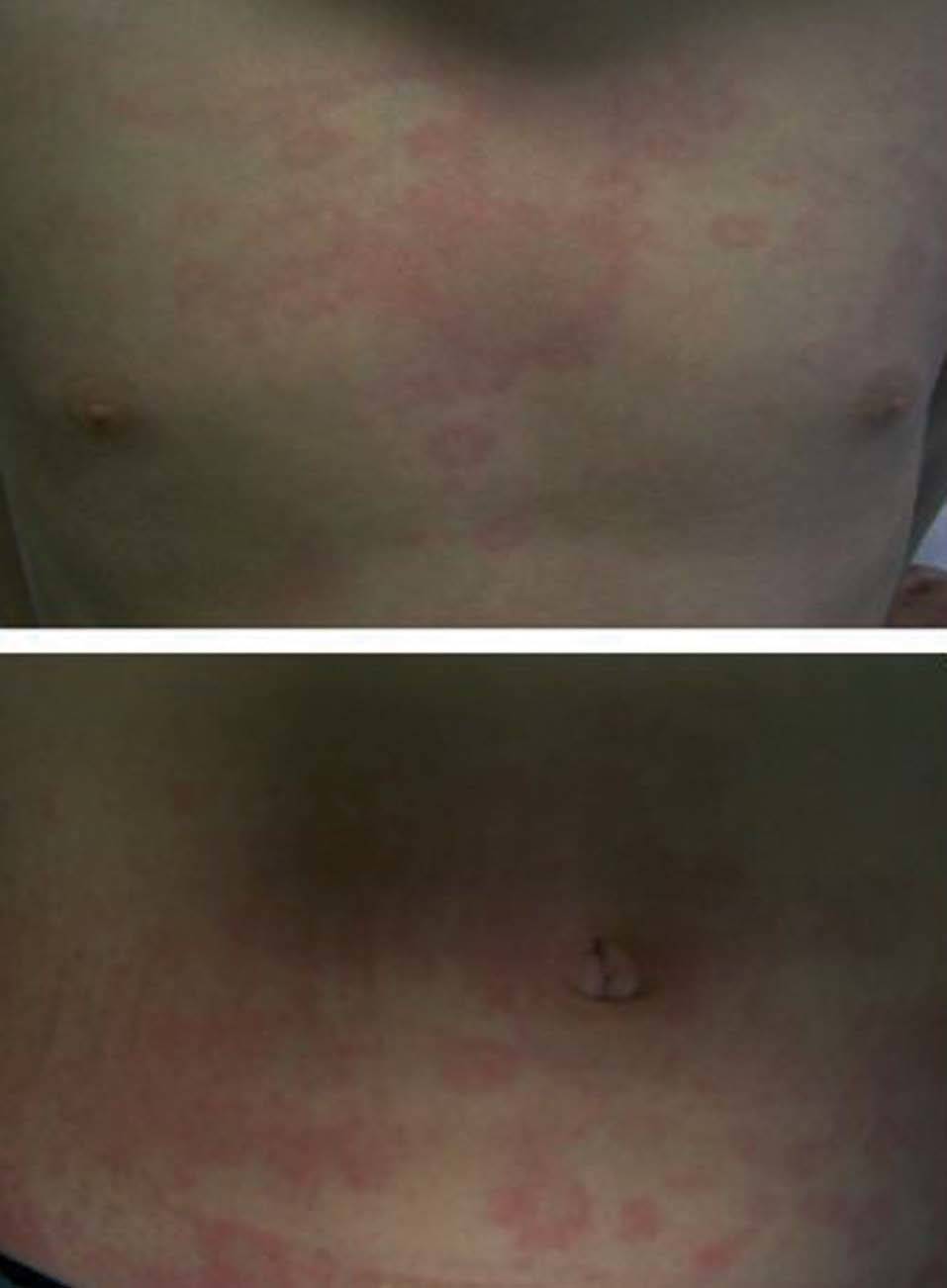
Las manifestaciones osteomusculares y las cutáneas son los rasgos clínicos distintivos de los episodios inflamatorios en el TRAPS y están presentes en la práctica totalidad de los pacientes. Dichas manifestaciones se caracterizan por la aparición de intensas mialgias en grupos musculares aislados, que son típicamente migratorias y cuya intensidad varía a lo largo del episodio inflamatorio4,11,12,74. Dichas mialgias son debidas a una fascitis monocítica inflamatoria, no a una miositis, razón por la cual las concentraciones séricas de creatinina y aldolasa suelen estar en valores normales76. Las mialgias suelen acompañarse de exantema eritematoso, habitualmente maculopapular, caliente y doloroso a la palpación, que se caracteriza por ser migratorio en sentido centrífugo y suele adquirir una imagen semejante a una diana conforme el episodio inflamatorio evoluciona en el tiempo (fig. 3)11,74. Las artralgias habitualmente están presentes durante los episodios agudos, acompañando a las manifestaciones musculares y cutáneas, mientras que la artritis, bien monoarticular u oligoarticular, se presenta en un porcentaje inferior de episodios74.
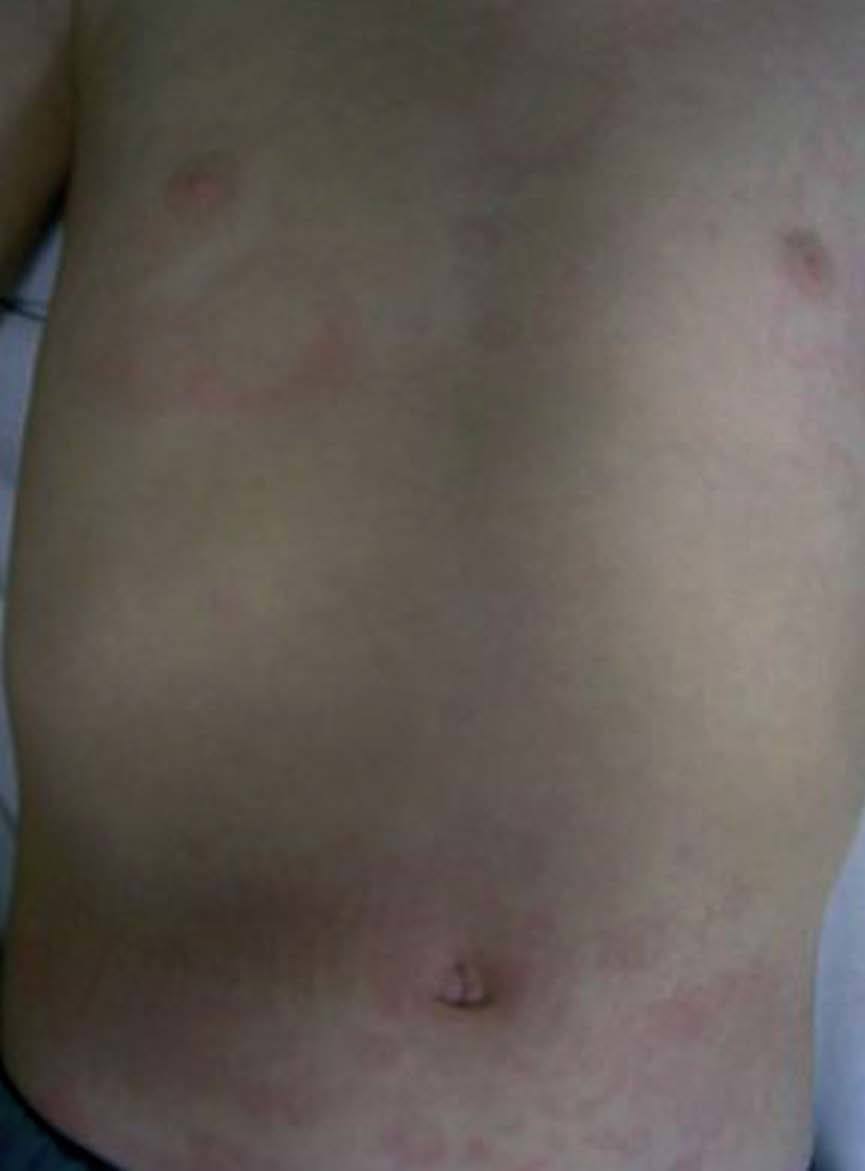
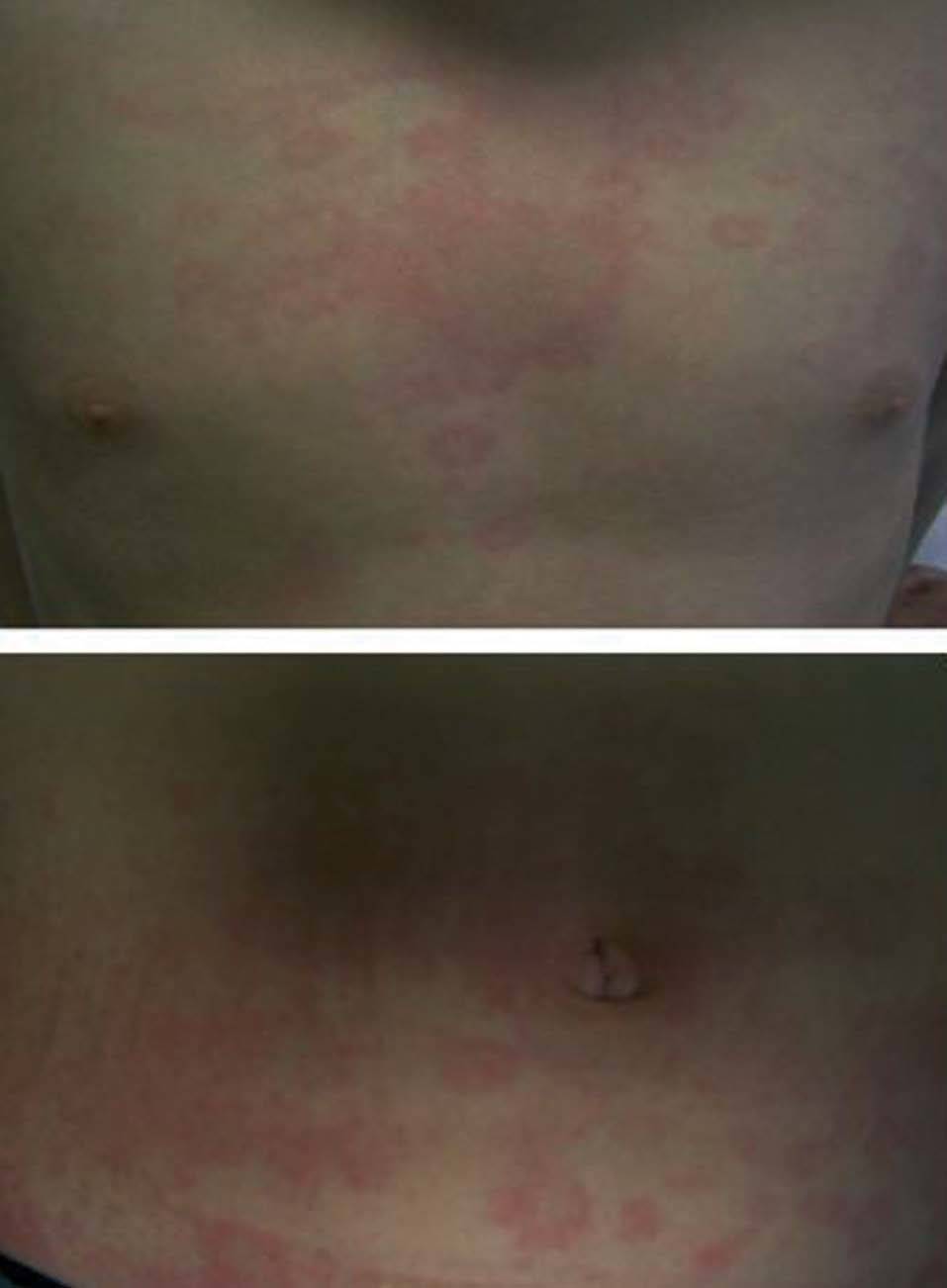
Fig. 3. Exantema eritematoso maculopapular y migratorio en el tórax y abdomen de un paciente afectado de síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAPS). (Imagen cortesía del Dr. Antón, Hospital Sant Joan de Déu.)
Las manifestaciones oculares son muy frecuentes (> 80%), habitualmente como conjuntivitis, edema y dolor periorbitales, tanto de forma unilateral como bilateral. Alguno de estos síntomas se ha identificado como prodrómico de los episodios inflamatorios en algunos pacientes con TRAPS67,74. Otras manifestaciones inflamatorias oculares, como uveítis o papiledema, son infrecuentes en los afectados de TRAPS.
Las manifestaciones digestivas están presentes en un elevado porcentaje de pacientes con TRAPS (hasta en el 92%) y habitualmente se presentan como dolor abdominal, que puede ser secundario bien a una peritonitis inflamatoria, bien a una inflamación de los músculos de la pared abdominal74. Es frecuente la asociación de vómitos y estreñimiento, y en algunas ocasiones los pacientes pueden precisar cirugía abdominal ante la presencia de un abdomen agudo, que suele ser debido a una peritonitis inflamatoria estéril.
Entre las manifestaciones menos frecuentes cabe destacar las adenopatías, el dolor torácico, bien sea por afectación pleural o por afectación de los músculos intercostales, y el dolor o la hinchazón testiculares.
La complicación más grave del TRAPS es la aparición de amiloidosis secundaria, habitualmente renal, como consecuencia de procesos inflamatorios repetidos a lo largo de años. Se ha descrito que los pacientes con TRAPS portadores de mutaciones que afectan a aminoácidos cisteína presentan un riesgo de desarrollo de la enfermedad mayor que los portadores de mutaciones en otros aminoácidos (el 24 frente al 2%, respectivamente)74.
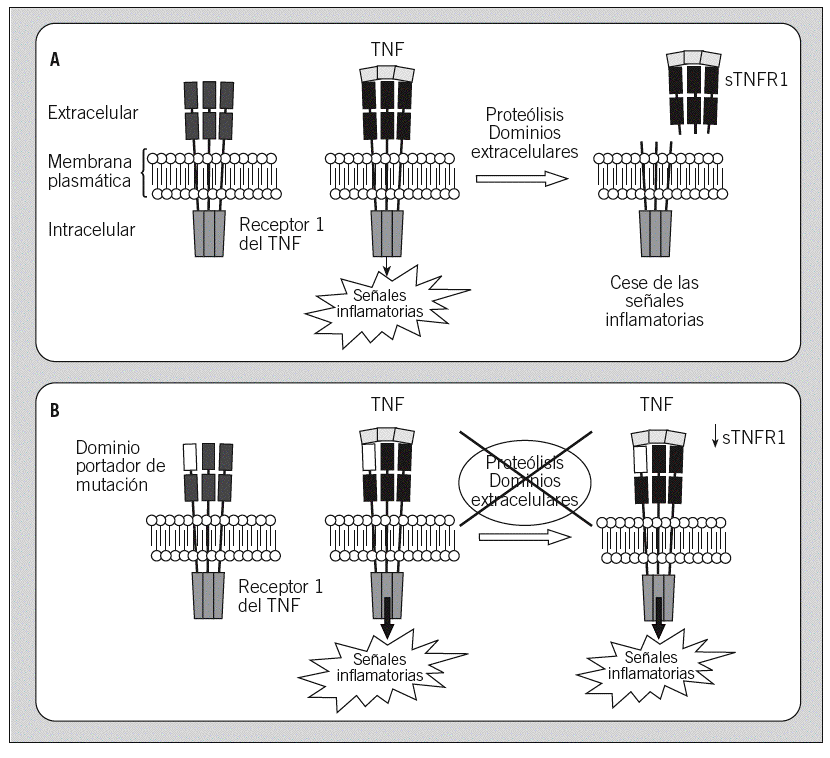
No hay ningún parámetro de laboratorio que sea específico o patognomónico del TRAPS. Durante los episodios agudos pueden apreciarse leucocitosis, neutrofilia, trombocitosis y cierto grado de anemia, así como una intensa reacción de fase aguda, con elevación de la VSG y de proteínas tales como la proteína C reactiva, haptoglobina, fibrinógeno, ferritina y proteína SAA1. En algunos pacientes se ha observado que durante los intervalos asintomáticos se encuentran disminuidos los valores plasmáticos de la forma soluble del receptor 1 del TNF (sTNFR1), mientras que durante los episodios inflamatorios agudos dichos valores alcanzan el intervalo considerado normal, pero no los valores que aparecen en un individuo sano en una situación inflamatoria semejante. Así pues, parece observarse un defecto en la generación del sTNFR1 en algunos de los pacientes con TRAPS1,3,74.
Genética. Era conocido que este síndrome tenía una herencia autosómica dominante, debido a la existencia de grandes familias con diversos miembros afectados en las que la enfermedad afectaba a todas las generaciones. Las bases moleculares del TRAPS se establecieron en 1999 al identificarse mutaciones en el gen TNFRSF1A, que codifica para el receptor 1 del TNF (también denominado p55 y CD120a)1. En concreto, dichas mutaciones se encontraron en una de las 2 copias del gen, es decir, en heterocigosis, tal como sería de esperar en una enfermedad dominante. Las mutaciones afectaban a aminoácidos que se localizan en los dominios extracelulares de la proteína y, si bien no afectan a la unión del receptor con su ligando (TNF), sí parecen afectar al mecanismo de proteólisis que regula la transducción de la señal inflamatoria a través de dicho receptor. Desde entonces se han identificado más de 50 mutaciones en dicho gen asociadas a enfermedad, la mayoría de las cuales se localizan en los exones 2, 3 y 4 del gen (fig. 1C)25. Asimismo, conforme se ha ido extendiendo el estudio genético como herramienta diagnóstica, ha aumentado el número de casos esporádicos identificados en los que la enfermedad es debida a la aparición de mutaciones de novo, por primera vez en ese paciente.
Ninguna de las mutaciones asociadas al TRAPS se encuentra en la población control sana. Sin embargo, hay 2 variantes del gen TNFRSF1A, denominadas Pro-46-Leu (P46L) y Arg-92-Gln (R92Q), que requieren un comentario especial. Ambas variantes se consideran en la actualidad mutaciones de baja penetrancia debido a que: a) están presentes en la población control sana en un bajo porcentaje (frecuencia alélica del 1,04%)74; b) en el seno de una familia dichas mutaciones pueden detectarse tanto en afectados de fiebre periódica como en individuos sanos, y c) los afectados de fiebre periódica y portadores de dicha mutación presentan unos síntomas clínicos heterogéneos en comparación con los que se observan en los pacientes con TRAPS clásico.
Etiopatogenia. En cuanto al mecanismo fisiopatológico subyacente en el TRAPS, no existe una respuesta clara. Se sabe que las mutaciones no afectan a la unión del receptor con su ligando y que no son mutaciones que generen un receptor activo de forma continua e independiente del ligando1. Se han propuesto varios modelos que están pendientes de demostración experimental. De ellos, el más aceptado es el defecto del shedding del receptor, según el cual las mutaciones del gen provocarían cambios en la proteína que afectan el shedding, es decir, la degradación proteolítica de los dominios extracelulares del receptor, que son el mecanismo homeostático natural de control de la señal inflamatoria a través de este receptor1. Dicho modelo también sería capaz de explicar por qué los pacientes con TRAPS presentan una alteración en la producción de sTNFR1 (fig. 4).
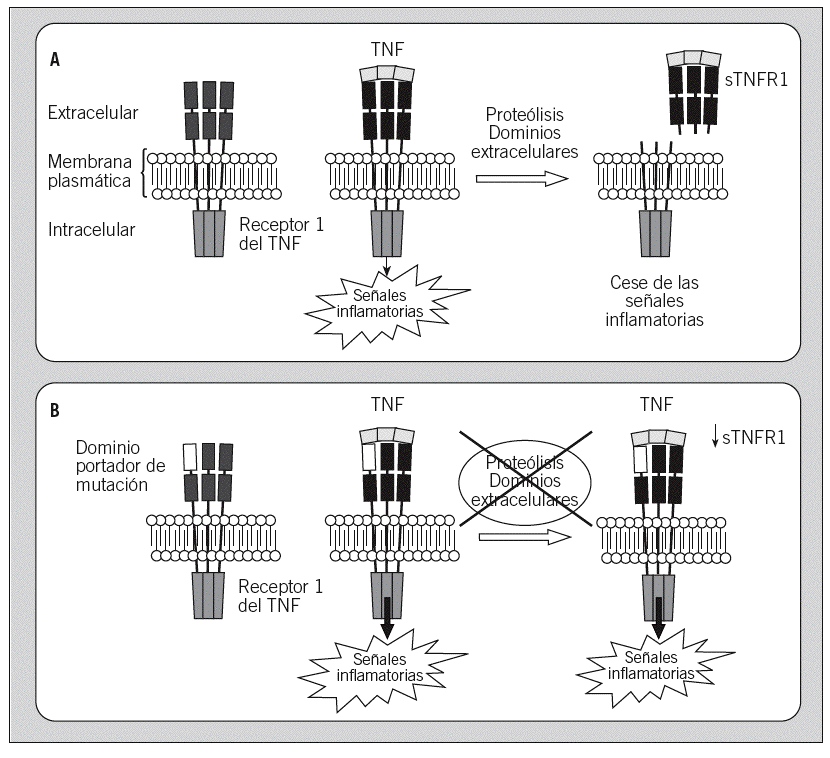
Fig. 4. Esquema de la transducción de la señal inflamatoria a través del receptor 1 del factor de necrosis tumoral (TNF) en un individuo sano (A) y en un paciente afectado de síndrome periódico asociado al receptor del TNF (TRAPS) y portador de una mutación en el receptor 1 del TNF (B). sTNFR1: forma soluble del receptor 1 del TNF.
Tratamiento. Los pacientes con TRAPS no responden a la colchicina y los antiinflamatorios no esteroideos, y presentan una buena respuesta, desde el punto de vista clínico y de laboratorio, a los glucocorticoides a dosis altas11,74. En pacientes en edad pediátrica se recomienda el empleo puntual de glucocorticoides para abortar los episodios inflamatorios agudos, mientras que en pacientes adultos, o en pacientes pediátricos que no responden a los glucocorticoides, se recomienda el uso de etanercept, agente biológico bloqueador del TNF11,74,77,78. A pesar de su eficacia para controlar los síntomas clínicos, este fármaco no es capaz de normalizar de forma continua los parámetros de inflamación subclínica, y se ha descrito algún caso aislado de desarrollo de amiloidosis secundaria, a pesar del buen control de los síntomas clínicos con etanercept70,79. En este sentido, se recomienda un estrecho seguimiento de la función renal, digestiva o tiroidea en estos pacientes, dado que son los lugares más comúnmente afectados en la amiloidosis secundaria. Recientemente se ha comunicado la buena respuesta clínica y de laboratorio de un caso aislado de TRAPS con el agente bloqueador de la interleucina 1 anakinra, con lo que se abre la posibilidad del uso terapéutico de estos fármacos en el síndrome TRAPS80.
Conclusiones
Las enfermedades autoinflamatorias sistémicas agrupan una serie de entidades clínicas diferentes, que comparten bases fisiopatológicas comunes, de reciente descripción. Dentro de dicho grupo de enfermedades, los HPFS constituyen su principal subgrupo. En los estudios llevados a cabo en pacientes españoles se han identificado casos de todos y cada uno de estos HPFS, a semejanza de lo que ocurre en otras poblaciones europeas. La baja frecuencia de todos y cada uno de estos síndromes, junto con la falsa idea de que sólo afectan a individuos de determinados grupos étnicos, ha hecho que todos estos síndromes se hayan pasado por alto o minusvalorado en el diagnóstico diferencial de pacientes aquejados de fiebre de origen desconocido. Asimismo, el solapamiento de signos y síntomas entre ellos, así como la ausencia de marcadores hematológicos o bioquímicos específicos, ha dificultado y retrasado tanto su diagnóstico definitivo como la instauración de un tratamiento adecuado. La disponibilidad actual de estudios genéticos de los genes responsables de cada uno de estos síndromes permite la confirmación de la sospecha clínica, así como la instauración de un tratamiento personalizado, sobre la base del mecanismo fisiopatológico subyacente, y posibilita asimismo una correcta evaluación del curso de la enfermedad y un adecuado consejo genético.
Agradecimiento
Los autores desean dar las gracias a los pacientes, a sus familiares y a sus médicos por la colaboración y el estímulo constante, que han hecho posible la presente revisión.
