







Inflammatory bowel disease (IBD) is a disorder of unknown aetiology that provokes chronic inflammation of the gastrointestinal tract. Anti-tumour necrosis factor drugs have represented a major advance in the treatment of IBD patients in the last few years and also have a good safety profile. Nevertheless, these treatments are not effective in all patients and, in initial responders, there can be a loss of response in the long-term. Consequently, new treatments are needed for IBD, aimed at distinct therapeutic targets.
In the last few years, new molecules have been incorporated into the therapeutic armamentarium of IBD patients. Golimumab is an anti-tumour necrosis factor monoclonal antibody with demonstrated effectiveness in the treatment of ulcerative colitis. The use of CT-P13 (biosimilar infliximab) has been approved in Europe for the same indications as the original infliximab. More recently, vedolizumab, an anti-α4β7 integrin monoclonal antibody, has been approved for the treatment of Crohn's disease and ulcerative colitis. A large number of molecules are currently under development, some of which will, in the future, broaden the therapeutic options available in the treatment of IBD patients.
Finally, in the next few years, studies should aim to identify factors predictive of response to the distinct biological agents for IBD in order to allow personalised selection of the best therapeutic alternative for each patient.
La enfermedad inflamatoria intestinal (EII) es un trastorno de etiología desconocida consistente en una inflamación crónica del tubo digestivo. Los fármacos dirigidos contra el factor de necrosis tumoral han representado un hito en el tratamiento de los pacientes con EII en los últimos años contando además con un buen perfil de seguridad. No obstante, estos tratamientos no son eficaces en todos los pacientes y, en aquellos que responden inicialmente, se ha descrito una pérdida de respuesta a lo largo del tiempo. Por estos motivos, es necesario el desarrollo de nuevos tratamientos para la EII, dirigidos hacia diferentes dianas terapéuticas.
En los últimos tiempos se han incorporado nuevas moléculas al arsenal terapéutico de los pacientes con EII. El golimumab es un anticuerpo monoclonal que se dirige contra el factor de necrosis tumoral y que ha demostrado ser eficaz en el tratamiento de la colitis ulcerosa. Asimismo, ha sido aprobado en Europa el uso de CT-P13 (infliximab biosimilar) para las mismas indicaciones que el infliximab original. Más recientemente, vedolizumab, un anticuerpo monoclonal dirigido frente a las integrinas α4β7 ha sido aprobado para el tratamiento de la enfermedad de Crohn y la colitis ulcerosa. En la actualidad se están desarrollando un gran número de moléculas, algunas de las cuales vendrán, en un futuro, a ampliar las opciones terapéuticas en los pacientes con EII.
Finalmente, en los próximos años los estudios deberán ir dirigidos a identificar factores predictores de respuesta a los distintos fármacos biológicos para la EII con el fin de seleccionar, de forma más personalizada, la mejor alternativa terapéutica para cada paciente.
Inflammatory bowel disease (IBD) is a chronic disorder of unknown aetiology that involves a pathological response of both the innate and acquired immune system, leading to chronic inflammation of the gastrointestinal (GI) tract. It is the result of the interaction of various factors, including genetic susceptibility, environmental factors, infectious agents, commensal enteric flora and immune disorders.1,2 The wide range of factors involved in the development of the disease, together with the complexity of the immune system, present multiple therapeutic targets, which are reflected in the large diversity of molecules that have been evaluated as potential treatments for IBD.
The era of biological therapy in the treatment of IBD began in 1998, when the United States Food and Drug Administration approved infliximab for the treatment of patients with Crohn disease.3–5 Many new biological drugs have been developed and approved since then, all of which have so far targeted tumour necrosis factor-alpha (TNF). Anti-TNF drugs have been a milestone in the treatment of patients with IBD in recent years, reducing the need for surgery and hospital admission and, most importantly, improving quality of life; they also have a good safety profile. Nevertheless, these treatments are not effective in all patients, and loss of response over time has been described in initial reponders.6–8 Furthermore, although they are safe in general, they are associated with adverse effects and are expensive.
For these reasons, new treatments with a more specific mechanism of action directed at different therapeutic targets must be developed for IBD, in order to achieve a more local effect in the inflamed organ.
Three new molecules have recently been added to the IBD therapeutic arsenal: golimumab and CT-P13 (infliximab biosimilar), which are anti-TNF drugs, and vedolizumab, which inhibits lymphocyte migration to the tissues of the GI tract by blocking the α4β7 integrin.
The aim of this review is to make a practical, critical appraisal of the biological molecules recently approved for the treatment of IBD, their characteristics, dosage, indications and safety profile. Finally, we will briefly present some of the groups of molecules being developed for the treatment of IBD.
GolimumabGolimumab (Simponi, Janssen Biotech, Inc., Horsham, PA, United States) is an alternative anti-TNF monoclonal antibody for patients with ulcerative colitis (UC) refractory to conventional treatment. Golimumab was approved in the United States by the Food and Drug Administration in May 2013; more recently, in October 2013, the European Medicines Agency (EMA) approved golimumab for the treatment of patients with UC who have been refractory or intolerant to conventional treatments.
Mechanism of actionGolimumab is a transgenic monoclonal antibody (immunoglobulin G1) that specifically targets an epitope on the TNF molecule.9,10 It binds to both soluble and transmembrane TNF with even stronger affinity than adalimumab and infliximab.11 It is superior to infliximab and adalimumab in terms of conformational stability and ability to inhibit TNF-induced cellular cytotoxicity and human endothelial cell activation.11
Like infliximab and adalimumab, golimumab acts by inducing apoptosis of inflammatory cells by binding to transmembrane TNF. Contrary to what might be expected of a full-length IgG such as golimumab, it is less effective than infliximab and adalimumab in inducing apoptosis via transmembrane TNF. For this reason, it could be less effective than adalimumab and infliximab in granulomatous diseases (such as Crohn disease [CD]), in which the effect due to the interaction of the antibody with the transmembrane TNF is very important.11 Finally, as the serum golimumab concentration necessary to neutralise the soluble TNF is lower than in the case of other anti-TNFs, golimumab can be administered every 4 weeks.
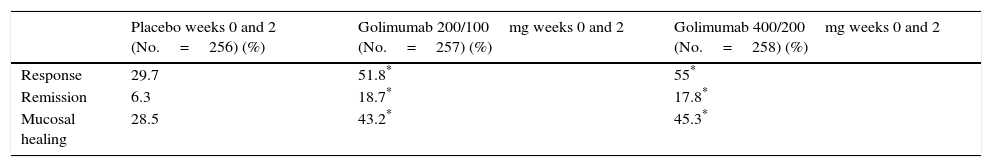
Efficacy of golimumab in ulcerative colitisThe PURSUIT-SC induction study evaluated the efficacy of golimumab in inducing remission in patients with moderate-severe UC refractory or intolerant to treatment with thiopurines, steroids or aminosalicylates.12 The study included a phase 2 dose-finding and a phase 3 dose-confirmation substudy, and included patients with UC with a Mayo score between 6 and 12, with at least 2 points on the endoscopic subscore. Patients were randomised to receive placebo, golimumab 100mg/50mg (before dose selection only), golimumab 200mg/100mg or golimumab 400mg/200mg subcutaneously at weeks 0 and 2.
The primary endpoint was to evaluate the week-6 response after dose selection. The secondary endpoints were week-6 clinical remission, mucosal healing and quality of life. Although all patients were included in the safety analysis, patients randomised to receive golimumab 100mg/50mg were not considered, as this dose was not included in the phase 2 substudy. Data from 774 patients were eventually analysed. All the study endpoints were reached for patients treated with golimumab compared with the placebo group (Table 1).
Efficacy outcomes for golimumab in the induction of response in patients with moderate-severe ulcerative colitis at week 6 (PURSUIT-SC induction study).
| Placebo weeks 0 and 2 (No.=256) (%) | Golimumab 200/100mg weeks 0 and 2 (No.=257) (%) | Golimumab 400/200mg weeks 0 and 2 (No.=258) (%) | |
|---|---|---|---|
| Response | 29.7 | 51.8* | 55* |
| Remission | 6.3 | 18.7* | 17.8* |
| Mucosal healing | 28.5 | 43.2* | 45.3* |
No.: number of patients.
The PURSUIT-maintenance study evaluated the efficacy of subcutaneous golimumab in maintaining remission in patients with moderate-severe UC.13 Patients who had presented a response in the induction studies with golimumab were included. Patients who had responded to induction treatment with golimumab were randomised to receive placebo, golimumab 50mg or golimumab 100mg every 4 weeks until week 54. Patients who had received placebo and responded received maintenance placebo. Finally, patients who had not achieved a response with golimumab or placebo received golimumab 100mg every 4 weeks. The study endpoint was the response over the 54 weeks. The secondary endpoints, evaluated at weeks 30 and 54, were clinical remission and mucosal healing.
Of the patients who initially responded to golimumab (464), the percentage of patients in remission at week 54 was significantly higher in the groups that received golimumab (50mg and 100mg) compared to the group that received placebo (Table 2).
Efficacy outcomes for golimumab in the maintenance of response (week 52) in patients with moderate-severe ulcerative colitis (PURSUIT-maintenance study).
| Placebo/4 weeks (No.=156) (%) | Golimumab 50mg/4 weeks (No.=153) (%) | Golimumab 100mg/4 weeks (No.=154) (%) | |
|---|---|---|---|
| Response | 31.4 | 47* | 50.6* |
| Remission | 15.4 | 23.5** | 28.6* |
| Mucosal healing | 26.9 | 41.8*** | 43.5* |
Golimumab is presented in the form of pre-filled syringes containing 0.5mL of solution with 50mg of golimumab.
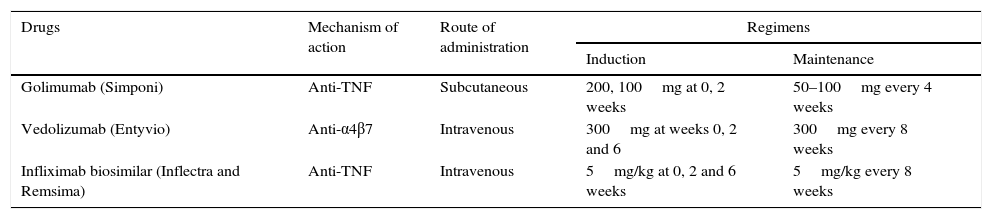
Recommended dose of golimumab for the treatment of ulcerative colitisAdministration of 200mg of golimumab subcutaneously at week 0 and 100mg at week 2 is recommended, followed by 50mg every 4 weeks thereafter for maintenance. For patients weighing 80kg or more, administration of 100mg every 8 weeks as maintenance is recommended (Table 3).
New drugs available for inflammatory bowel disease, mechanisms of action, route of administration and recommended doses.
| Drugs | Mechanism of action | Route of administration | Regimens | |
|---|---|---|---|---|
| Induction | Maintenance | |||
| Golimumab (Simponi) | Anti-TNF | Subcutaneous | 200, 100mg at 0, 2 weeks | 50–100mg every 4 weeks |
| Vedolizumab (Entyvio) | Anti-α4β7 | Intravenous | 300mg at weeks 0, 2 and 6 | 300mg every 8 weeks |
| Infliximab biosimilar (Inflectra and Remsima) | Anti-TNF | Intravenous | 5mg/kg at 0, 2 and 6 weeks | 5mg/kg every 8 weeks |
Golimumab is indicated for the treatment of moderate-severe UC in adults who have had a poor response to conventional treatment, including corticosteroids and immunosuppressants, or who have intolerance or contraindications to these therapies.
SafetyIn the PURSUIT-SC induction study, the percentage of adverse effects was similar in patients exposed to golimumab and in those who received placebo.12 The most common adverse events among patients treated with golimumab were headache and nasopharyngitis. Serious adverse effects were similar in both the golimumab and placebo groups (3.0% vs 6.0%). The percentage of serious infections was also similar in both groups (0.5% vs 1.8%). One patient in the golimumab 400mg/200mg group died due to postoperative complications, while another patient in the same group presented a demyelinising disorder. The percentage of patients who had to discontinue treatment due to adverse effects was low (0.5% in those treated with golimumab and 0.9% in those who received placebo). The proportion of patients with injection site reactions was low (3.4% in the golimumab groups and 1.5% in the placebo group), with no differences as regards the golimumab dose.
In the PURSUIT-maintenance study, golimumab showed a good safety profile.13 The rates of adverse effects, serious adverse events, infections and serious infections were similar among the different treatment groups. Likewise, the rate of treatment dropout due to side effects was similar in the 3 study groups. The percentage of patients with injection-site reactions was similar in the groups treated with golimumab and in the placebo group.
VedolizumabVedolizumab (Entyvio, Millennium Pharmaceuticals, Cambridge, MA, United States) is a humanised monoclonal antibody that binds specifically to the α4β7 integrin heterodimer, selectively blocking lymphocyte migration to the intestine, without affecting their migration to other organs such as the central nervous system (CNS). During the development of this drug, the molecule underwent several modifications, receiving different names (LDP02 and MLN0002) until the current version, known as MLN0002 or vedolizumab.14
Mechanism of actionIBD is a chronic inflammation in the GI tract necessitating the migration of inflammatory mediator cells to the affected tissues. The migration of leukocytes to the inflamed intestinal tissues is regulated by complex molecular mechanisms. Integrin α4β7 is a glycoprotein that is activated on the surface of the circulating B and T lymphocytes, and interacts with mucosal vascular addressin cell adhesion molecules (MAdCAM-1) in the endothelial venules. MAdCAM-1 is selectively expressed on the endothelium of the intestinal venules, binding the lymphocytes that circulate at high velocity in the blood stream.15 Thus, these lymphocytes migrate from the endothelial surface to the lamina propria and the tissues. Persistence of excessively high levels of these lymphocytes in the intestinal tissues is part of the pathophysiological process of IBD.16 Several studies have observed a significant increase in the expression of α4β7 and MadCAM-1 in the colon of patients with UC compared to patients with irritable bowel syndrome (IBS).17
Natalizumab, the predecessor of vedolizumab, showed the potential of this mechanism of action in phase 2 studies. Natalizumab is a monoclonal antibody that has been shown to be effective in both multiple sclerosis and CD. It binds to the α4 subunit of the integrins, thus blocking the interactions of the α4β1 and α4β7 heterodimers with their receptors, including fibronectin, VCAM-1 and MadCAM-1.18–20 Despite its efficacy, natalizumab was withdrawn from the market in 2005 due to the onset of progressive multifocal leukoencephalopathy (PML), which is a serious CNS infection associated with treatment with this drug. It was reintroduced in 2006 for use in surveillance programmes and is currently approved in the United States (although not in Europe) for CD. Fifty-two thousand patients have received natalizumab as monotherapy since its reintroduction, most for the treatment of multiple sclerosis; 10 new cases of PML have been reported.21 PML is believed to be due to reactivation of the John Cunningham virus in immunosuppressed states, facilitating dissemination of the virus through the bloodstream. By binding to integrin α4β1, natalizumab blocks lymphocyte migration to the CNS, thus preventing clearance of the virus from the CNS by the immune system. The difference between vedolizumab and natalizumab is that the latter blocks lymphocyte migration to multiple organs (like the GI tract and CNS), while vedolizumab acts specifically on the GI tract. Studies in primates have shown that vedolizumab is specific to the GI tract and is not associated with changes in the cerebrospinal fluid.22 Furthermore, in a study conducted in a small group of healthy volunteers who underwent lumbar puncture before and after a 450mg dose of vedolizumab, no differences were observed in CD4+, CD8+ or the CD4:CD8 ratio.23
A monoclonal antibody against α4β7 was first used in 1996, demonstrating its capacity to control colitis in animal models.24 Tamarind monkeys in captivity spontaneously developed chronic colitis that resembled UC. The monkeys included in the study were diagnosed with colitis using endoscopic and histological criteria, and randomised to receive the anti-α4β7 drug or a non-therapeutic monoclonal antibody by intramuscular administration. Primates that received the anti-α4β7 antibody presented rapid clinical, endoscopic and histological improvement that was not observed in the untreated animals.24
Efficacy of vedolizumabThe first study with an anti-α4β7 in humans was published in 2000 as an abstract. It was a double-blind, placebo-controlled study that included 29 patients with moderate-severe UC. Patients were randomised to receive humanised antibody (LDP02) in ascending doses: 0.15mg/kg subcutaneously, 0.15mg/kg intravenously, 0.5mg/kg intravenously and 3mg/kg intravenously or placebo. A single 0.5mg/kg dose was sufficient to completely saturate the antibody receptors.25
In a phase 1 study conducted in patients with UC, Feagan et al.25 demonstrated that, after a single infusion of vedolizumab (then called MLN0002), blockade of the α4β7 integrin was higher than placebo in the clinical and endoscopic induction. The first phase 2 study was carried out in patients with UC naive to biological treatment. A total of 181 patients with UC were randomised to receive MLN0002 at doses of 2mg/kg, 0.5mg/kg or placebo at weeks 0 and 4.26 The primary outcome measure was remission at week 6. In both groups (2 and 0.5mg/kg), the percentages of clinical remission were higher in patients treated with MLN0002 than in those exposed to placebo (33%, 32% and 14%, respectively, p=0.03). Similarly, the percentage of patients who reached endoscopic remission was significantly higher in those treated with MLN0002 (2 and 0.5mg/kg) than with placebo (28%, 12% and 8%, respectively).26
A second phase 2 study included 185 patients with CD naive for anti-TNF treatment who were randomised to receive MLN0002 at doses of 2mg/kg, 0.5mg/kg or placebo at weeks 0 and 4.27 At week 6, 37% and 30% of patients treated with 2 and 0.5mg/kg of MLN0002, respectively, achieved remission, compared to 21% of patients treated with placebo. The percentage of adverse effects was similar among groups.27
Despite the efficacy of the anti-α4β7 antibody demonstrated in these studies, approximately 40% of patients developed antibodies against the drug.26 Furthermore, an antibody titre greater than 1: 125 was associated with a lower likelihood of response due to lower saturation of the α4β7 integrin.26 For this reason, Millennium Pharmaceuticals developed a new antibody formulation (the initial formulation was produced by mouse myeloma cell line N0 and the new one by a cell system based on Chinese hamster ovary), which was evaluated in a dose-finding study (2, 6 and 10mg/kg).28 In this study, the serum vedolizumab (MNL0002) concentration increased proportionally with the dose administered; the α4β7 receptors on peripheral blood lymphocytes were saturated at all the doses evaluated. Later, in an open-label phase 2 study that included patients with CD and UC, the new formulation was evaluated at the same doses (2, 6 and 10mg/kg) but administered more frequently (at weeks 0, 2 and 6 instead of weeks 0 and 4, as in the first phase 2 studies).29 Only 4% of patients in this study developed antibodies to vedolizumab. The authors concluded that the combination of change of formulation, higher dose and more frequent administration of the drug contributed to less antibody formation.
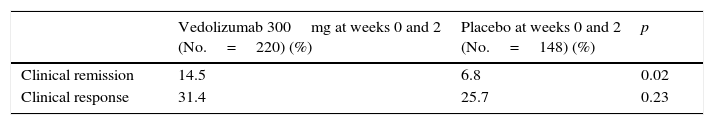
Vedolizumab has been more recently evaluated in phase 3 studies (GEMINI I, II, III and LTS).30–33 The GEMINI I study evaluated the efficacy of vedolizumab in the induction and maintenance of remission in patients with moderate-severe UC.30 Clinical response was defined as a reduction in the Mayo Clinic score of at least 3 points and a decrease of at least 30% with respect to baseline, with a decrease of at least 1 point on the “rectal bleeding” subscale or an absolute “rectal bleeding” score of ≤1. Clinical remission was defined as a Mayo Clinic score of ≤2, with a score of ≤1 in all the subscores. In the induction phase, 374 patients were randomised to receive vedolizumab 300mg at weeks 0 and 2 or placebo (cohort 1), while 521 patients received open-label vedolizumab 300mg at weeks 0 and 2 (cohort 2). In the maintenance study, patients who had responded to vedolizumab at week 6 were randomised to continue on vedolizumab 300mg/8 weeks, vedolizumab 300mg/4 weeks or placebo until week 52.
The percentage of patients who presented a response at week 6 was higher in patients treated with vedolizumab than in those who received placebo (47% vs 25.5%). Furthermore, the percentage of patients who achieved remission and the percentage of patients with mucosal healing were higher in patients treated with vedolizumab than in those who received placebo (Table 4).
Efficacy outcomes for vedolizumab in the induction of remission (week 6) in patients with ulcerative colitis (GEMINI I study).
| Vedolizumab 300mg at weeks 0 and 2 (No.=225) (%) | Placebo at weeks 0 and 2 (No.=149) (%) | p | |
|---|---|---|---|
| Clinical response | 47.1 | 25.5 | < 0.01 |
| Clinical remission | 16.9 | 5.4 | < 0.01 |
| Mucosal healing | 40.9 | 24.8 | < 0.01 |
No.: number of patients.
Source: Feagan et al.30
At week 52, the percentage of patients in remission was significantly higher in the vedolizumab group compared to those randomised to placebo. Moreover, the percentage of patients with a sustained clinical response, mucosal healing and steroid-free remission was significantly higher in patients randomised to vedolizumab than in those who received placebo. In the post hoc analysis, the vedolizumab regimen of 300mg/4 weeks was no better than the regimen of 300mg/8 weeks. Concomitant treatment with steroids or immunosuppressants was not associated with greater drug efficacy. Additionally, previous anti-TNF treatment failure was not associated with lower efficacy of vedolizumab therapy.
A correlation between vedolizumab levels and response was observed. The mean vedolizumab concentration with the 300mg/4 weeks dosing schedule was higher than in patients with 300mg/8 weeks (38.3 vs 11.2 (g/mL)). However, in both cases, saturation of the α4β7 integrin on T lymphocytes was greater than 95%.
A small number of patients (3.7%) developed antibodies to vedolizumab. Concomitant treatment with immunosuppressants reduced the incidence of formation of antibodies to vedolizumab.
The GEMINI II study evaluated the efficacy of vedolizumab in the induction and maintenance of remission in patients with CD refractory to conventional treatments or to anti-TNF.31 It included patients with moderate-severe CD defined as a Crohn's disease activity index (CDAI) score of 220–450 together with one of the following: elevated C-reactive protein (CRP), or endoscopic activity (≥3 large ulcers or ≥10 aphthous ulcers) or faecal calprotectin ≥250μg/g plus evidence of ulcers on the computed tomography, magnetic resonance, intestinal transit or capsule endoscopy. The primary endpoints for the induction phase were clinical remission (defined as a CDAI ≤150 at week 6) and clinical response (defined as a decrease of ≥100 points with respect to the CDAI at week 0).
In the induction phase, 368 patients were randomised to receive vedolizumab 300mg or placebo at weeks 0 and 2 (cohort 1), while 747 patients received open-label vedolizumab 300mg at weeks 0 and 2. At week 6, the percentage of patients who achieved remission was significantly higher in the vedolizumab group than in the placebo group. However, the percentage response was similar in both groups (Table 5).
Efficacy outcomes for vedolizumab in the induction of remission (week 6) in patients with Crohn disease (GEMINI II study).
| Vedolizumab 300mg at weeks 0 and 2 (No.=220) (%) | Placebo at weeks 0 and 2 (No.=148) (%) | p | |
|---|---|---|---|
| Clinical remission | 14.5 | 6.8 | 0.02 |
| Clinical response | 31.4 | 25.7 | 0.23 |
At week 6, the 461 patients who had presented a response with vedolizumab treatment were randomised to receive vedolizumab 300mg/4 weeks, vedolizumab 300mg/8 weeks or placebo until week 52. The primary endpoint of the maintenance phase was clinical remission at week 52 (CDAI ≤150). At week 52, the percentage remission in patients who received vedolizumab 300mg, both every 4 and every 8 weeks, was significantly higher than in the placebo group (Table 6).
Efficacy outcomes for vedolizumab in the maintenance of remission (week 52) in patients with Crohn disease (GEMINI II study).
| Vedolizumab 300mg/4 weeks (No.=154) (%) | Vedolizumab 300mg/8 weeks (No.=154) (%) | Placebo (No.=153) (%) | p | |
|---|---|---|---|---|
| Clinical remissiona | 36.4 | 39 | 21.6 | < 0.05 |
| Clinical responseb | 45.5 | 43.5 | 30.1 | < 0.05 |
| Steroid-free remission | 28.8 | 31.7 | 15.9 | < 0.05 |
| Sustained remissionc | 16.2 | 21.4 | 14.4 | NS |
No.: number of patients; NS: not statistically significant.
Clinical response was defined as a decrease in the CDAI ≥100 points at week 52 with respect to baseline.
Sustained clinical remission was defined as a CDAI ≤150 at 80% of the study visits, including the last one (week 52).
Source: Sandborn et al.31
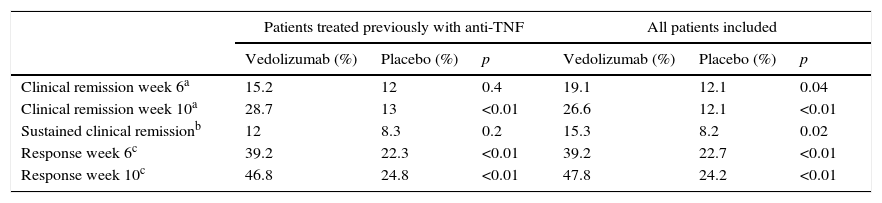
The GEMINI III study is a randomised, double-blind, placebo-controlled study that evaluated the safety and efficacy of vedolizumab in remission induction in 416 patients with CD refractory to other treatments.34 The primary endpoint, which was to demonstrate the efficacy of vedolizumab in inducing remission at week 6 in patients with previous anti-TNF treatment failure, was not reached in this study. However, the clinical response at week 6 (decrease in CDAI ≥100 points with respect to the baseline score) and remission at week 10 were greater in patients treated with vedolizumab (Table 7).
Efficacy of vedolizumab in the induction of remission in patients with Crohn disease (GEMINI III study).
| Patients treated previously with anti-TNF | All patients included | |||||
|---|---|---|---|---|---|---|
| Vedolizumab (%) | Placebo (%) | p | Vedolizumab (%) | Placebo (%) | p | |
| Clinical remission week 6a | 15.2 | 12 | 0.4 | 19.1 | 12.1 | 0.04 |
| Clinical remission week 10a | 28.7 | 13 | <0.01 | 26.6 | 12.1 | <0.01 |
| Sustained clinical remissionb | 12 | 8.3 | 0.2 | 15.3 | 8.2 | 0.02 |
| Response week 6c | 39.2 | 22.3 | <0.01 | 39.2 | 22.7 | <0.01 |
| Response week 10c | 46.8 | 24.8 | <0.01 | 47.8 | 24.2 | <0.01 |
Clinical response was defined as a decrease in the CDAI ≥100 points with respect to baseline.
Source: Sands et al.34
Finally, an open-label extension study is currently underway to evaluate the long-term safety of vedolizumab (GEMINI LTS).35 Preliminary results support its long-term safety in the treatment of CD and UC. The full study results (at 7 years of follow-up) will be available in 2016.
Mode of presentationThe packs contain 300mg of vedolizumab powder for concentrate for solution for infusion.
PosologyThe recommended dose regimen of Entyvio is 300mg administered by intravenous infusion at 0, 2 and 6 weeks and then every 8 weeks thereafter (Table 3). Treatment discontinuation should be assessed in patients who have not presented a response after 14 weeks of treatment. In patients who lose the initial response over time, the frequency of administration may be increased to 300mg/4 weeks.
IndicationsVedolizumab is indicated in adults with moderately to severely active CD or UC who have had an inadequate response with, lost response to or are intolerant to conventional treatment or to an anti-TNF.
SafetyIn general, the percentage of side effects in patients treated with vedolizumab was similar to those described in the placebo group. No cases of PML have been described. Nevertheless, future studies will provide information on the long-term safety profile of vedolizumab.
CT-P13The price of biological drugs, together with their increasingly widespread use, presents a dilemma due to the costs associated with these treatments. This has led several national agencies to restrict their use when the patient has been in remission for some time.36,37 The development of generic versions of small-molecule drugs has enabled price reductions of 80% compared to the original branded product.38 As with other drugs, when the patents for biologics are approaching their expiry dates, other companies begin to develop “copies” of the original molecules, which in this case are known as biosimilars. The development of biosimilar drugs will make biological drugs similar to the originals available at a lower cost.
A biosimilar is a copy of an approved biological drug for which the patent has expired.39 However, while a generic drug is an exact copy of a small, relatively simple molecule, the biosimilar is not identical to the original molecule; instead it may differ due to differences in the manufacturing process, type of cell culture used for synthesis, purification process, formulation or storage conditions. These changes could account for differences in the quality, efficacy and safety of treatment, especially due to changes in the immunogenicity.40 This does not only occur with biosimilars, since different batches of the original biologics can present differences due to changes in the manufacturing process over time. As a result of these changes in the production process, the manufacturers of the original biologics sometimes have to demonstrate their comparability to the regulatory authorities.41
The name “biosimilar” was proposed by the EMA, which was the first regulatory authority to establish the process for approval of these drugs. Following approval of the first biosimilar drug by the EMA in 2005, 16 biosimilars have been authorised to date in Europe.
The process for approving a biosimilar drug is different from that followed for the development of an original drug.41 The EMA establishes that the manufacturer of the biosimilar molecule has to demonstrate comparability with the original in 3 aspects: physico-chemical characteristics of the molecules, non-clinical comparability (evaluating the theoretical mechanisms of action in different models) and clinical comparability, which should be done for the indication in which the patient population is most sensitive.42 Despite small differences, the process is largely the same in Japan, Australia, Canada and the United States.
After a complex evaluation of the data provided by the manufacturer of the biosimilar molecule, the product will be accepted if it is considered “highly similar”. Once this similarity has been demonstrated, the requirements for clinical studies are less stringent than for the reference product, assuming that the high similarity also predicts that the clinical results will be highly similar.42
Comparability of CT-P13 with RemicadeIn 2013, the EMA approved the first biosimilar of Remicade (infliximab), developed by Celltrion as CT-P13 and marketed as Remsima by Celltrion and Inflectra by Hospira.42
Clinical studies with this drug were conducted in rheumatoid arthritis (RA) and ankylosing spondylitis (AS).43,44 In addition to these indications, the EMA approved the use of CT-P13 for all the indications of Remicade (which is known as extrapolation).42
CT-P13 has an amino acid sequence identical to that of Remicade, as this is an indispensable requisite for a biosimilar. The physico-chemical characterisation of CT-P13 showed that the product is highly similar to Remicade. The different functions of CT-P13 evaluated, including the affinity to bind to TNF, its suppression of cytokine production in several human cell lines and the other analyses carried out, showed results within the parameters described for Remicade. The only difference observed between Remicade and CT-P13 was in the proportion of afucosylated forms, which in the latter translated into a lower binding affinity to the FcγRIIIa receptor, and lower antibody-mediated cellular cytotoxicity in the natural killer cells in in vitro experiments. However, when these experiments were conducted in more physiological conditions, the differences disappeared, with both Remicade and CT-P13 showing similar cytotoxic activity.42
The development of the first infliximab biosimilar gave rise to considerable debate about the requirements necessary for the approval of a biosimilar product in all the indications of the reference product. European guidelines establish that, if properly justified, the biosimilar should receive authorisation for all the indications of the reference product, which is known as extrapolation of indications.42
Extrapolation is justified because the objective of studies for the development of biosimilars is not to demonstrate efficacy in the different indications per se, which has already been demonstrated by the original drug, but that the similarity demonstrated in the clinical, preclinical and pharmacokinetic studies means that comparable clinical outcomes are highly likely.
Several scientific societies have taken a stand against extrapolation, as they consider that clinical data are necessary to determine the efficacy of the biosimilar in all indications.45,46 Others, in contrast, have supported extrapolation, and have opted for strict post-marketing surveillance.47 Detractors of extrapolation of indications to IBD argue that the differences in afucosylation could alter the effect of the drug on this disease. In fact, differences have been observed between RA and IBD as regards the response to treatment with other monoclonal antibodies: some drugs have been shown to be effective in some diseases and not in others, while others have been shown to be active in both diseases but at different doses.45,46
Efficacy of CT-P13 in clinical studiesStudies for the approval of CT-P13 were conducted in patients with RA and AS. The aim of the PLANETRA study was to compare the efficacy and safety of Remicade with CT-P13 in patients with RA.43 The primary endpoint was to evaluate the American College of Rheumatology 20% (ACR20) response rate at week 30. CT-P13 showed an efficacy similar to Remicade at week 30, with a similar pharmacokinetic and immunogenicity profile. CT-P13 was well tolerated, with a safety profile comparable to Remicade.
The aim of the PLANETAS study was to compare the pharmacokinetics, efficacy and safety of Remicade with CT-P13 in patients with AS.44 The primary endpoint was to evaluate the area under the drug concentration curve and the maximum concentration between weeks 22 and 30 of treatment. The response and safety of both treatments were evaluated as secondary endpoints. The study showed that the pharmacokinetic profiles of Remicade and CT-P13 were similar in patients with AS. CT-P13 was well tolerated, with a safety and efficacy profile comparable to Remicade, at least up to week 30.
One of the issues raised by detractors of extrapolation of indications of CT-P13 is the suitability of the diseases chosen for the clinical studies. Patients with RA are the largest patient group who receive treatment with anti-TNF drugs, but from the point of view of obtaining approval from the regulatory authorities, they do not appear to be the most sensitive population.
In order to obtain drug approval, a risk management plan must be provided for each biosimilar antibody. In addition to the usual pharmacovigilance, the EMA management plan for CT-P13 includes several post-authorisation extension studies that in total will include more than 6000 patients, one of these being a randomised clinical trial comparing the efficacy of CT-P13 with Remicade in patients with CD. Although the EMA has put forward solid arguments to justify extrapolation of the Remicade indications, the results of a clinical trial comparing the efficacy of CT-P13 and Remicade in CD will support and facilitate acceptance of the use of the biosimilar in IBD.
Molecules under development for the treatment of inflammatory bowel diseaseResearch into the pathophysiological mechanisms involved in IBD has at the same time led to the discovery of potential therapeutic targets for the development of new drugs for the treatment of this disease. Most molecules under investigation block T lymphocyte activation or leucocyte adhesion, or inhibit the production or effect of proinflammatory cytokines.
Blockade of proinflammatory cytokinesTumour necrosis factorTNF-kinoid is a vaccine that aims to induce production of neutralising polyclonal antibodies to TNF by the patient himself. A phase 1–2 open-label study in patients with CD evaluated 3 doses of TNF-kinoid in 22 patients.48 No serious adverse events were observed and all patients completed the study. The preliminary results of the phase 2 trial were not so encouraging.49
HMPL-004Andrographis paniculata is considered to be a medicinal plant. Extracts of this plant have been reported to have anti-inflammatory properties by inhibiting nitric oxide synthesis and the NF-κβ pathway.50 It has also been shown to have an inhibitory effect on the activation of the cells of the immune system.
HMPL-004 is one of the components of Andrographis paniculata extract that has shown promising results in the treatment of IBD. This molecule is currently being evaluated in phase 3 studies in CD and UC.
Interleukins 12 and 23Interleukins 12 and 23 are proinflammatory cytokines that share a common subunit (p40). Ustekinumab is an IgG1 monoclonal antibody that binds to the p40 subunit of interleukins 12 and 23, and has been shown to be effective in the treatment of psoriatic arthritis and psoriasis.51–53The efficacy of ustekinumab in the induction of remission in patients with CD was first evaluated in a double-blind, placebo-controlled study by Sandborn et al.54 The patient group treated with ustekinumab presented a higher percentage of clinical response at weeks 4 and 6 than those treated with placebo; nevertheless, no differences were observed at week 8.
The efficacy of ustekinumab in the induction and maintenance of remission in patients with CD was subsequently evaluated by Sandborn et al. in a phase 2b study.55 Patients with moderate-severe CD refractory to anti-TNF treatment were included. Patients were randomised to receive intravenous ustekinumab 1, 3 or 6mg/kg or placebo at week 0. The clinical response at week 6 was significantly greater in the ustekinumab group compared to placebo. However, no differences in remission were observed. It should be noted that, although the primary endpoint was established at week 6, a higher percentage of patients treated with ustekinumab achieved response or remission at week 8 than at week 6, showing that the mechanism of action is slower and that week 6 is perhaps too early to assess the response.
Patients who responded to ustekinumab at week 6 were randomised again to receive subcutaneous ustekinumab 90mg every 8 weeks or placebo.55 At week 22, the percentage of patients with clinical response and remission was significantly higher in the ustekinumab group compared to placebo. Phase 3 studies of this molecule for the treatment of CD are currently underway.
For now, physicians can only use ustekinumab as “off label” treatment in patients with CD. The recommended dose for remission induction in patients with CD is not well established; the most widely used regimen at present is 90mg subcutaneously at weeks 0, 1, 2 and 3, followed by maintenance treatment of 90mg subcutaneously every 8 weeks thereafter.
Few side effects were reported, and were comparable to those reported in the placebo group in clinical trials for CD. Nevertheless, data from patients with psoriasis show an excellent safety profile, with rare, mild side effects.
Other interleukinsBoth interleukin-13 and interleukin-6 are proinflammatory cytokines that have been tested as potential therapeutic targets in IBD. To date, the results of studies with drugs targeting these molecules have been modest.56,57 New molecules against these cytokines are currently being evaluated for the treatment of IBD.58
Administration of anti-inflammatory cytokinesThe administration of anti-inflammatory cytokines could be a therapeutic approach in the treatment of IBD. Anti-inflammatory cytokines that have been evaluated as possible treatments in patients with IBD are interleukin-10, interleukin-11 and interferon-β.59–66 Unfortunately, these cytokines have not been shown to be useful in the treatment of IBD in the studies conducted to date.
Leucocyte adhesion blockadeEtrolizumab is a humanised monoclonal antibody against the β7 subunit of integrins α4β7 and αEβ7. The EUCALYPTUS study is a phase 2, randomised, double-blind, placebo-controlled clinical trial that evaluated the efficacy of etrolizumab in 124 patients with moderate-severe UC who were randomised to receive etrolizumab 100mg at weeks 0, 4 and 8 or etrolizumab 420mg (loading dose) and then 300mg at weeks 2, 4 and 8 or placebo.67 The percentage of patients who achieved clinical remission (Mayo Clinic Score ≤2 with no subscore >1) was 21% in the etrolizumab 100mg group, 8% in the etrolizumab 300mg group and 0% in the placebo group (p=0.004 and p=0.05, respectively, compared with placebo). Phase 3 studies are currently underway to confirm these promising results.
Chemokines constitute a complex family of proteins recently identified as chemotactic cytokines that can activate different cell types in the immune system; they differ from classic chemoattractants in their specificity for particular leucocyte sub-types. These molecules are involved in the recruitment and migration of leukocytes to the intestinal mucosa. Some studies have suggested that aberrant expression of chemokine receptor 9, which is expressed in the small intestine and colon, could be a therapeutic target in CD.68,69
Vercirnon is an oral inhibitor of chemokine receptor 9 that has been evaluated in a double-blind, placebo-controlled clinical trial in patients with CD. Patients were randomised to receive 250mg/24h, 250mg/12h, 500mg/24h or placebo for 12 weeks. The primary endpoints were the response at weeks 8 and 52. In the induction phase, only the group who received 500mg/day presented a percentage response significantly higher than placebo. At week 52, 47% of patients who received vercirnon presented remission, compared to 31% in the placebo group.70 Phase 3 studies to evaluate the efficacy of this molecule in IBD are currently underway.
Blockade of cytokine-mediated signal cascadesJanus kinase inhibitorsThe involvement of the Janus kinase (JAK) family JAK1 and JAK3 in the process of signal transduction of interleukin-2 and interleukin-6 receptors (such as interleukin-12 and interleukin-23) has made the inhibition of JAKs a potential therapeutic target in IBD. Tofacitinib is an inhibitor of JAK1, JAK2 and JAK3 that can modulate the signals of a large group of proinflammatory cytokines, such as interleukins 2, 4, 7, 9, 15 and 21. These integrins participate in lymphocyte activation, proliferation and functions.
A phase 2 randomised, placebo-controlled clinical trial evaluated the efficacy of tofacitinib in patients with moderate-severe UC.67 Patients were randomised to receive 4 different doses of tofacitinib or placebo for 8 weeks. In this study, only the group who received the highest dose (15mg/12h) presented a significantly higher response than the placebo group (78% vs. 42%). The percentage of patients with mucosal healing was 18% in the group who received 3mg, 30% in the 10mg group, 27% in the 15mg group and 2% in the placebo group.67 The drug safety profile was good. Patients who received tofacitinib had increased low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol due to an unexplained mechanism, as it theoretically only acts on the JAK expressed specifically in the cells of the immune system. A phase 3 study is currently underway to evaluate the efficacy and safety of tofacitinib in patients with UC.
LaquinimodLaquinimod is a small synthetic molecule that—by a mechanism that has not been fully identified—transforms the T cells into an anti-inflammatory phenotype, decreasing the production of proinflammatory cytokines.71 In a phase 2, double-blind, placebo-controlled, dose-finding study, patients were randomised to receive 0.5, 1, 1.5 or 2mg of laquinimod daily or placebo for 8 weeks. Surprisingly, the highest percentages of remission and response were obtained in the group of patients treated with the lowest dose of laquinimod.72
MasitinibThe mastocytes of the GI tract are sentinels of the immune system that are located in the mucosa and submucosa of the digestive tract in the barrier between the host and the environment. The activation and proliferation of mastocytes can be observed in certain processes, including IBD. Masitinib is a potent selective tyrosine kinase inhibitor that is targeted at the c-kit receptor (expressed in mastocytes), but also modulates other cytokines.73 A phase 2 study is currently underway in patients with CD.
ConclusionsNew molecules have recently been incorporated into the therapeutic arsenal of patients with IBD. Golimumab is a monoclonal antibody against TNF that has been shown to be effective in the treatment of UC. In Europe, approval of CT-P13 (infliximab biosimilar) for the same indications as the original infliximab has marked the debut of an anti-TNF molecule that can substantially reduce the cost of biological treatment. More recently, vedolizumab, a monoclonal antibody against the α4β7 integrin, has been approved for the treatment of CD and UC. This mechanism of action of this drug, which selectively prevents GI inflammation, differs from that of classic anti-TNFs.
Furthermore, the study of the pathophysiology of IBD has led to the identification of numerous therapeutic targets. A large number of molecules are currently under development, some of which will be used in the future to extend the therapeutic options in patients with IBD.
Finally, in the coming years, studies should be aimed at identifying factors that can predict the response to different biological drugs for IBD, in order to select the best therapeutic alternative for each patient on a more individual basis.
Conflict of interestsM. Chaparro: conferences, support for research and/or training activities: MSD, AbbVie, Hospira, Dr. Falk Pharma. J.P. Gisbert: scientific consultancy, conferences, support for research and/or training activities: MSD, AbbVie, Takeda, Hospira, Kern Pharma, Pfizer, Janssen, Ferring, Faes Farma, Shire Pharmaceuticals, Chiesi, Laboratorios Casen Fleet, Otsuka Pharmaceutical, Uriach and Dr. Falk Pharma and Gebro Pharma.
Please cite this article as: Chaparro M, Gisbert JP. Nuevas moléculas en el tratamiento de la enfermedad inflamatoria intestinal. Gastroenterol Hepatol. 2016;39:411–423.
