




INTRODUCCIÓN
El cáncer colorrectal (CCR) corresponde a la segunda neo plasia más común y a la segunda causa más frecuente de muerte relacionada con cáncer en España1. Se asume que el CCR está causado por una compleja interacción entre los factores de susceptibilidad existentes en cada individuo y los factores de riesgo presentes en el ambiente. Teniendo en cuenta las variaciones mundiales de la incidencia del CCR, la dieta correspondería al factor de riesgo ambiental más importante, mientras que los factores genéticos serían clave en la predisposición individual.
PREDISPOSICIÓN GENÉTICA AL CÁNCER COLORRECTAL
La genética tiene un papel principal en la predisposición al CCR y en su iniciación y progresión. Una de las razones por las que esta neoplasia proporciona un modelo para comprender la genética del cáncer es la facilidad para observar los diferentes estadios del desarrollo del tumor. Esta secuencia bien definida y conocida desde hiperplasia o proliferación aberrante en la cripta hasta adenoma benigno, carcinoma in situ y, finalmente, carcinoma, ha permitido determinar las mutaciones somáticas implicadas en cada caso. Así, se ha visto que estas mutaciones ocurren en un determinado orden, primero aparecen las que afectan al gen APC, seguidas de las que afectan a la familia de los genes RAS, mientras que los cambios en TP53 tienden a aparecer más hacia el final de la secuencia2,3.
El CCR se ha divido tradicionalmente en esporádico y familiar o hereditario, dependiendo de si existe o no algún tipo de agregación familiar. En un reciente estudio multicéntrico, prospectivo y de ámbito nacional realizado por nuestro grupo, se valoró que el grupo de CCR hereditario/familiar y esporádico son un 29,4 y un 70,6% del total, respectivamente4. Entre los casos familiares de CCR, los atribuibles a síndromes hereditarios de alta penetrancia serían sólo un pequeño porcentaje, mientras que el resto correspondería a los causados por factores genéticos de baja penetrancia, pero mucho mayor prevalencia. Entre los síndromes hereditarios de alta penetrancia causantes de CCR más frecuentes, se encuentran el cáncer colorrectal hereditario no polipósico y la poliposis adenomatosa familiar, aunque juntos no llegan a totalizar un 5% del total de casos de CCR.
El cáncer colorrectal hereditario no polipósico (CCHNP) es una enfermedad hereditaria autosómica dominante y hasta el momento es la más común predisposición genética a CCR, a menudo con otras manifestaciones extracolónicas como neoplasias de endometrio, ovario, estómago, intestino delgado, sistema hepatobiliar y urinario, cerebro y piel (véase la revisión reciente de Chung y Rustgi5). El CCHNP es el 1-3% de los casos de cáncer colorrectal, dependiendo de la población estudiada. En la población española se estima que representa el 2,5%6. El CCHNP está asociado a mutaciones germinales en genes implicados en la vía de reparación de mal apareamiento del ADN (MMR, mismatch repair), específicamente MLH1, MSH2, MSH6 y PMS2. Las mutaciones en MLH1 y MSH2 son las mayoritarias y suponen alrededor del 90% de las mutaciones encontradas en familias CCHNP, mientras que mu taciones en MSH6 suponen alrededor del 7-10%, y mutaciones en PMS2 corresponden a menos del 5%. La penetración del CCR asociado a mutaciones en estos genes es del 80%, aproximadamente. También se han publicado mutaciones en los genes MSH3, EXO1 y TGFßR2 en algunas familias CCHNP, aunque su significación clínica no está bien establecida7.
POLIPOSIS ADENOMATOSA FAMILIAR CLÁSICA Y ATENUADA
La poliposis adenomatosa familiar (PAF) es un síndrome hereditario de predisposición genética al CCR, con una incidencia de 1/7.000-10.000, que se caracteriza por la aparición de cientos a miles de adenomas a lo largo de colon y recto, normalmente empezando en la adolescencia. Si no se practica una colectomía, uno o varios adenomas progresarán inevitablemente hacia adenocarcinoma, habitualmente antes de finalizar la cuarta década de la vida. La proporción de CCR debido a PAF está alrededor del 1% del total o inferior. La penetrancia de este síndrome es prácticamente del 100% y se presenta comúnmente con lesiones gastroduodenales asociadas como poliposis glandular fúndica, adenomas o pólipos hiperplásicos, y es muy frecuente la existencia de lesiones hiperpigmentadas de la retina (véase la revisión reciente de Cruz-Correa y Giardiello8).
La PAF se hereda de forma autosómica dominante y la causan mayoritariamente las mutaciones en el gen supresor de tumores APC9. Todos los individuos portadores de una mutación germinal en este gen, que corresponde al primer golpe de la teoría de progreso del cáncer de Knudson, desarrollan PAF. El desarrollo de tumores en la PAF corresponde a la aparición del segundo golpe en forma de mutación somática en APC en el tejido afectado. El gen APC tiene un tamaño considerable, se compone de 15 exones y codifica para una proteína de 2.843 aminoácidos que comprende varios dominios con funciones determinadas. La mayoría de las mutaciones germinales en APC corresponden a mutaciones que comportan una terminación prematura de la proteína codificada. Además, existe una cierta correlación genotipo-fenotipo entre la localización de la mutación en el gen y la presentación clínica. Así, la presencia de cientos a miles de adenomas va ligada a una mutación en los codones 169-1.600; la hipertrofia congénita del epitelio pigmentario, a una mutación en los codones 463-1.387, y el síndrome de Gardner, una variante de PAF caracterizada por osteomas, fibromas dérmicos y quistes epidérmicos asociados a la poliposis, a una mutación en los codones 1.403-1.57810.
La PAF es un ejemplo de cáncer hereditario en el que las variaciones alélicas de un mismo gen dan lugar a una diversidad en el fenotipo. Así, se ha descrito una variante denominada poliposis adenomatosa familiar atenuada que se caracteriza por un inicio más tardío y un menor número de pólipos localizados de manera preferente en el colon derecho, y cuya alteración molecular consistiría en la presencia de mutaciones en el extremo 5' y 3' del gen APC.
Mediante métodos de detección rutinarios, hasta un 70% de pacientes con PAF clásica y un 10% de pacientes con PAF atenuada presentan mutaciones germinales en el gen APC. Recientemente, se ha descrito mutaciones germinales en el gen MYH de pacientes con múltiples adenomas colorrectales sin mutación identificada en APC11, que presentan un patrón hereditario recesivo. Esta entidad se ha denominado poliposis asociada al MYH y puede llegar a ser hasta un 7,5% de los pacientes con PAF clásica y un 5-30% de los pacientes con PAF atenuada en los que se ha descartado mutaciones en el gen APC. En el caso de la PAF atenuada, los pacientes con mayor número de pólipos (> 30), sin evidencia de transmisión vertical, tienen mayor probabilidad de presentar mutaciones en el gen MYH (25-60%).
MYH Y EL SISTEMA DE REPARACIÓN DEL ADN POR ESCISIÓN DE BASES
Al-Tassan et al11 investigaron una familia británica en la que 3 hermanos presentaban adenomas colorrectales múltiples y CCR. La secuenciación total del gen APC en ADN germinal de 2 de los individuos afectados, junto con el análisis de haplotipos y de expresión génica, excluyeron un defecto genético en ese gen. El análisis de la inestabilidad de microsatélites en el ADN extraído de 11 tumores de esa familia también excluyó un posible defecto de la reparación de mal apareamiento del ADN. Sin embargo, el patrón de las mutaciones observadas en los tumores de esa familia facilitó la identificación del factor genético causante. En este sentido, la secuenciación completa de APC en cada uno de los 11 tumores reveló 18 mu taciones somáticas, 15 de las cuales eran transversiones G:C * T:A. Este tipo de mutaciones corresponde sólo a un 10% de las identificadas somáticamente en el gen APC, y las mutaciones de pérdida de la pauta de lectura y la pérdida de heterozigosis son lo que más frecuentemente causa la inactivación somática de APC en tumores colorrectales12. La comparación de lo encontrado en esa familia británica, con una base de datos de más de 800 mutaciones somáticas en APC en pacientes con CCR esporádico o asociado a PAF, confirmó el incremento altamente significativo de transversiones G:C * T:A en esta familia.
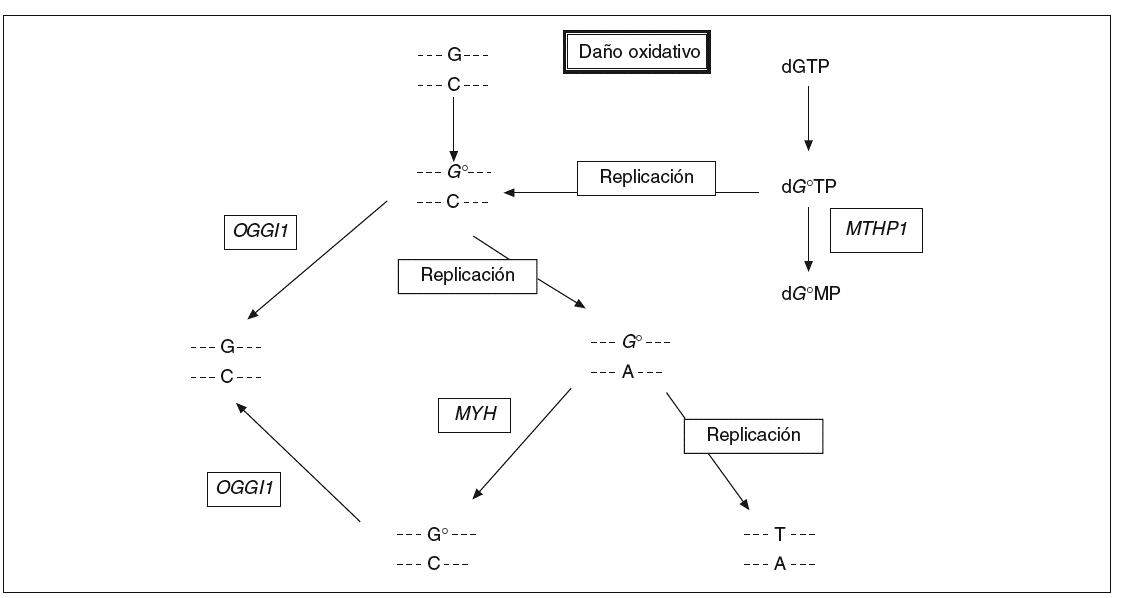
Los compuestos procedentes del oxígeno reactivo con un potencial dañino tienen un papel importante en procesos como el envejecimiento, el cáncer y las enfermedades neurodegenerativas. En humanos se estima que la frecuencia del daño oxidativo al ADN es de 10.000 lesiones por célula y día13. 8-oxo-7,8-dihidro2'deoxiguanosina (8-oxodG) es uno de los productos nocivos más estables del daño oxidativo al ADN y su concentración es alta en el cáncer de mama, pulmón y riñón (véase la revisión de Loft y Poulsen14). Este compuesto se aparea mal con residuos de adenina y causa las mutaciones G * T y C * A en la hebra naciente en la replicación del ADN (fig. 1). La incorporación de 8-oxodG en el ADN tiene lugar a través de la oxidación directa de los residuos de guanina en la cadena molde o gracias a la incorporación de 8-oxodG procedente del conjunto de nucleótidos libres. Las mutaciones aparecen después de la replicación del ADN debido a que las polimerasas incorporan adenosinas frente a 8-oxodG a un ritmo mucho mayor del que incorporan citosinas.
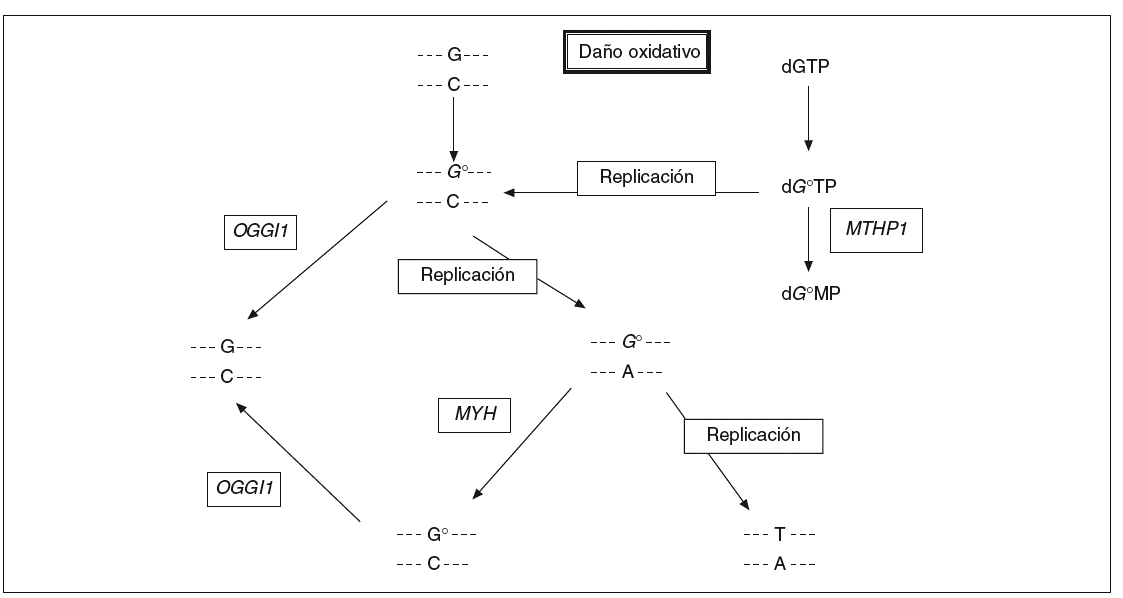
Fig. 1. La vía de reparación del ADN por escisión de bases. La proteína codificada por el gen MYH actúa en la cadena de ADN naciente eliminando las adenosinas mal apareadas con 8-oxodG.
En Escherichia coli existen 3 enzimas que ayudan a proteger a la célula contra los efectos mutagénicos de la oxidación de la guanina. La glucosilasa de ADN MutM elimina la base oxidada de los pares de base 8-oxodG:C en el ADN de doble cadena, la glucosilasa de ADN MutY escinde las adeninas incorporadas por error y apareadas con 8-oxodG durante la replicación, y la 8-oxodGTPasa MutT impide la incorporación de 8-oxodGTP en la cadena naciente de ADN en la replicación. Los genes homólogos de mutM, mutY y mutT en humanos han sido identi ficados y se denominan OGG115, MYH16 y MTH117, respectivamente.
Para determinar si un defecto heredado en la vía de reparación de 8-oxodG era la causa del patrón de mutaciones somáticas G:C * T:A en la familia estudiada, Al-Tassan et al11 secuenciaron por completo los genes OGG1, MYH y MTH1 en el ADN extraído de sangre de uno de los afectados. De esta forma se identificaron 2 mutaciones que cambian de forma no conservativa 2 aminoácidos del gen MYH (Y165C y G382D), mientras los demás hermanos afectados también eran hererozigotos compuestos para estas 2 mutaciones. El resto de los familiares no afectados eran heterozigotos para una de ellas o no portadores, lo que indica una transmisión de la enfermedad en esta familia con un patrón de herencia autosómico recesivo.
MYH, ¿UN GEN DE SUSCEPTIBILIDAD PARA EL CÁNCER COLORRECTAL?
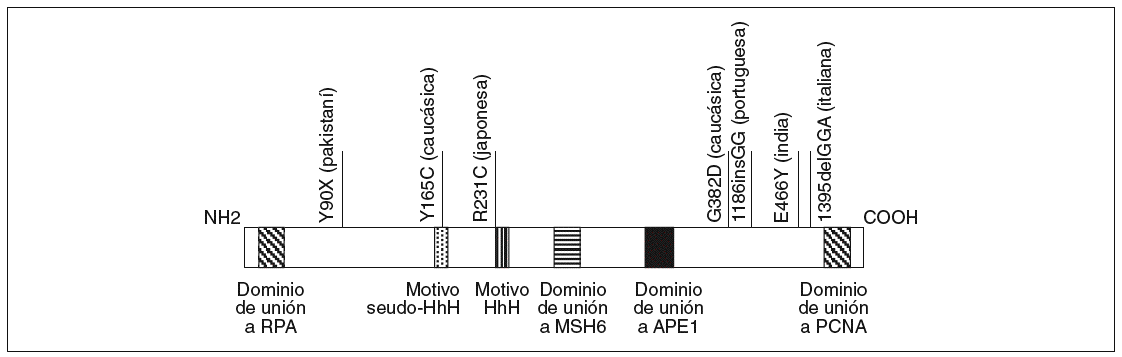
El gen MYH está localizado en el brazo corto del cromosoma 1 entre p32.1 y p34.3, tiene una longitud de 7.100 bases y contiene 16 exones que codifican una proteína de 535 aminoácidos con un 41% de homología con la proteína homóloga MutY de E. Coli16. Sus dominios funcionales han sido descritos previamente y se esquematizan en la figura 2.
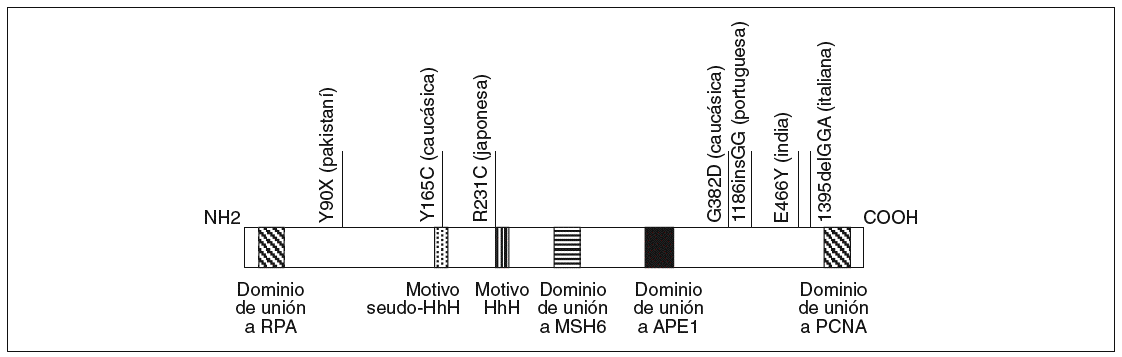
Fig. 2. Esquema de la proteína MYH. Se han resaltado los dominios funcionales definidos hasta el momento en la parte inferior y algunas mutaciones étnicas en la parte superior.
Al estudio de Al-Tassan et al11 han seguido otros que han caracterizado pacientes con adenomas colorrectales múltiples (generalmente entre 5 y 100 pólipos) sin mutaciones en el gen APC18-24. Estos estudios han permitido reafirmar el papel del gen MYH en lo que, como se ha comentado previamente, se ha empezado a denominar poliposis asociada a MYH, siguiendo una herencia autosómica recesiva. Otra cuestión importante detectada por los estudios mencionados ha sido la existencia de mutaciones étnicas o adscritas a un origen geográfico concreto (fig. 2). Así, las mutaciones G382D y Y165C corresponden a las mayoritariamente detectadas en la población caucásica, mientras que otras mutaciones se adscriben a poblaciones como la pakistaní18, la india18, la italiana21, la portuguesa23 o la japonesa25.
Si bien el papel del gen MYH en la poliposis colorrectal múltiple parece más que probado, estudios recientes han analizado su papel en la predisposición al CCR. Así, varios estudios de base poblacional han mostrado una clara asociación entre mutaciones bialélicas del gen MYH y el desarrollo de CCR, con una penetrancia del 100% a los 60 años26-28. En la mayoría de los casos, el CCR se acompaña de un número variable de pólipos sincrónicos (5-100). Sin embargo, cabe destacar que más de un 30% de los pacientes con mutaciones bialélicas que han desarrollado un CCR no presentan pólipos sincrónicos, por lo que la presencia de éstos como una posible aproximación para la detección de mutaciones es poco sensible26. La predisposición al CCR de los sujetos portadores de mutaciones monoalélicas en MYH es un tema actualmente controvertido. Se ha señalado un aumento del riesgo de CCR siguiendo un patrón autosómico dominante con base en la detección, en algunos estudios, de una mayor agregación familiar de CCR en estos sujetos29. Estudios más recientes han demostrado un aumento del riesgo de CCR en edades avanzadas en relación con la heterocigosis, lo que respalda la hipótesis de que el gen MYH actúa como un gen de susceptibilidad para el CCR de baja penetrancia26, aunque se requiere de estudios poblacionales mucho más amplios para corroborarla.
En conclusión, la poliposis asociada al gen MYH constituye un nuevo síndrome de herencia autosómica recesiva que predispone a los adenomas colorrectales al CCR, lo que supone un avance significativo en el conocimiento de los mecanismos genéticos implicados en esta neoplasia. La penetrancia de esta enfermedad, sus manifestaciones extracolónicas, el fenotipo de los heterocigotos, el tratamiento más adecuado y la posible existencia de alteraciones en otros componentes del sistema de reparación del ADN por escisión de bases son importantes aspectos aún por dilucidar.

