








La poliposis adenomatosa familiar (PAF) es una enfermedad hereditaria causada por variantes patogénicas del gen APC, que también se asocia con manifestaciones extracolónicas. El objetivo fue caracterizar las manifestaciones extracolónicas en una cohorte de pacientes con PAF clásica y la posible asociación genotipo-fenotipo.
Materiales y métodosEl diseño del estudio fue observacional descriptivo. Se recogieron las variables demográficas, clínicas y genéticas en función del tipo de variante patogénica (frameshift, nonsense, splicing, rearrangement y otras).
ResultadosSe incluyeron 45 pacientes con PAF (edad media 47años, rango 21-78; sexo femenino 51%), pertenecientes a 21 familias, con una mediana de 2 (rango 0-6) manifestaciones por paciente. El 80% (n=36) presentaron afectación del tracto digestivo superior, siendo los adenomas duodenales (73%), la poliposis fúndica (56%) y la presencia de ampuloma (36%) los hallazgos más frecuentes. Las afectaciones extraintestinales más frecuentes fueron el tumor desmoide (16%) y el carcinoma papilar de tiroides (13%). El 38% de los pacientes presentaron un fenotipo agresivo (SpigelmanIII-IV, displasia de alto grado, neoplasia invasiva, tumor desmoide y carcinoma papilar de tiroides). Las variantes patogénicas más habituales fueron frameshift (56%), nonsense (26%) y splicing (16%), localizadas principalmente en el exón15 (50%). No se demostró una correlación significativa entre el tipo de variante patogénica con la gravedad y la localización de las manifestaciones fenotípicas.
ConclusionesUna tercera parte de los pacientes con PAF presentan un fenotipo agresivo, sin demostrarse una correlación entre el tipo de alteración genética y las manifestaciones fenotípicas.
Familial adenomatous polyposis (FAP) is a hereditary disease caused by mutations in the APC gene, which is also associated with extracolonic manifestations. The objective was to characterize the extracolonic manifestations in a cohort of patients with classic FAP and the possible genotype-phenotype association.
Materials and methodsThe study design was observational and descriptive. Demographic, clinical, and genetic variables were collected based on the type of mutation (frameshift, nonsense, splicing, rearrangement, and others).
ResultsWe included 45 patients with FAP (mean age 47years, range 21-78; 51% female), belonging to 21 families, with a median of 2 (range 0-6) manifestations per patient. Eighty percent (n=36) had upper digestive tract involvement, with duodenal adenomas (73%), fundic gland polyposis (56%), and ampullary adenoma (36%) being the most frequent findings. The most common extraintestinal manifestations were desmoid tumors (16%) and papillary thyroid carcinoma (13%). Thirty eight percent of the patients presented an aggressive phenotype (SpigelmanIII-IV, high-grade dysplasia, invasive neoplasia, desmoid tumor, and papillary thyroid carcinoma). The most common genetic mutations were frameshift (56%), nonsense (26%), and splicing (16%), primarily located in exon15 (50%). No significant correlation was found between the type of genetic mutation and the severity or location of phenotypic manifestations.
ConclusionsOne-third of patients with FAP present an aggressive phenotype, without a demonstrated correlation between the type of genetic alteration and the phenotypic manifestations.
La poliposis adenomatosa familiar (PAF) es una enfermedad hereditaria causada por variantes patogénicas del gen APC1. Esta enfermedad se caracteriza por la aparición de cientos a miles de adenomas colorrectales, con un riesgo muy elevado de progresión a cáncer colorrectal si el paciente no recibe tratamiento de forma precoz2,3, así como una frecuente asociación con manifestaciones extracolónicas4. La colectomía profiláctica y un mejor seguimiento de estos pacientes han mejorado el pronóstico. En la actualidad, los tumores desmoides y los tumores del tracto gastrointestinal superior son las principales causas de morbimortalidad, por lo que es crucial poder estratificar el riesgo de estos pacientes5.
El gen APC es un gen supresor de tumores ubicado en el brazo largo del cromosoma5 (5q21) que genera un polipéptido de 2.844 aminoácidos con un papel importante en la adhesión celular, la transducción de señales y la activación transcripcional6,7. Las variantes de la línea germinal del gen APC siguen un patrón de herencia autosómico dominante y su penetrancia es de cerca del 100%. Hasta un tercio de los casos diagnosticados que no pertenecen a familias previamente identificadas representan variantes patogénicas de novo de la línea germinal o mosaicismos8. Se han descrito más de 3.000 variantes patogénicas diferentes en el gen9. Los puntos críticos donde las variantes generan una proteína truncada se localizan en el exón15, en los codones 1061 y 130910,11, con una penetrancia genética casi completa para las manifestaciones colónicas, pero variable para las manifestaciones extracolónicas12.
Dado que en estudios previos se ha descrito una posible correlación entre la localización de la variante patogénica y la penetrancia de las manifestaciones extraintestinales en los pacientes con PAF13,14, nos planteamos realizar un estudio descriptivo de las características fenotípicas extracolónicas y genéticas en los pacientes con PAF.
Material y métodosDiseño del estudioSe realizó un estudio observacional descriptivo de todos los pacientes con PAF controlados en un hospital terciario de referencia en España. Se recogieron a través de la historia clínica las siguientes variables: datos demográficos (edad y sexo), hallazgos endoscópicos (número de adenomas duodenales), adenomas duodenales mayores de 10mm, clasificación de Spigelman, presencia de ampuloma, desarrollo de poliposis fúndica proximal (presencia de 10 o más pólipos de glándula fúndica) y neoplasia gástrica, desarrollo de manifestaciones extraintestinales (tumor desmoide, osteoma, carcinoma papilar de tiroides) y resultados del análisis genético según el tipo y la localización de la mutación. El seguimiento y el diagnóstico de las manifestaciones extracolónicas se llevó a cabo según las medidas de cribado de neoplasias asociadas a poliposis colorrectales recomendadas por la Guía de práctica clínica sobre el diagnóstico y la prevención del cáncer colorrectal de la AEG15. Se definió como comportamiento más agresivo la presencia de un SpigelmanIII-IV, de adenoma duodenal/gástrico con displasia de alto grado, de adenoma duodenal/gástrico con neoplasia invasiva, de tumor desmoide y de carcinoma papilar de tiroides. Se estableció el tipo de variante patogénica de acuerdo a lo siguiente: a)frameshit: inserción o deleción de un número de nucleótidos que no es múltiplo de tres en una secuencia de ADN; b)nonsense: variante puntual en una secuencia de ADN que provoca la aparición de un codón de terminación prematuro; c)splicing: variante que inserta, elimina o cambia un número de nucleótidos en el sitio específico en el que tiene lugar el empalme durante el procesamiento del ARN mensajero; d)rearrangement: gran reordenamiento genético o configuración anormal de un fragmento largo de material cromosómico por alteraciones como la inversión o la translocación, y e)no disponible: por resultado no disponible en nuestra historia electrónica.
ObjetivoEl objetivo principal del estudio fue conocer la prevalencia de las manifestaciones extracolónicas y el tipo de variante patogénica, así como su correlación en pacientes con PAF.
Estudio estadísticoLos datos se recogieron en Microsoft Excel 2021 y se analizaron con el paquete estadístico para ciencias sociales (SPSS) versión 25 (SPSS para Windows, versión 25.0; SPSS Inc, Chicago, IL, EE.UU.). Se utilizaron tablas de frecuencia para las variables cualitativas y calculamos parámetros estadísticos simples, como la media, la mediana y la desviación estándar (DE) para las variables cuantitativas. Se empleó la prueba chi-cuadrado de Pearson para comparar proporciones entre grupos. Los resultados se presentaron en forma de medias ±DE, medianas, rangos o porcentajes.
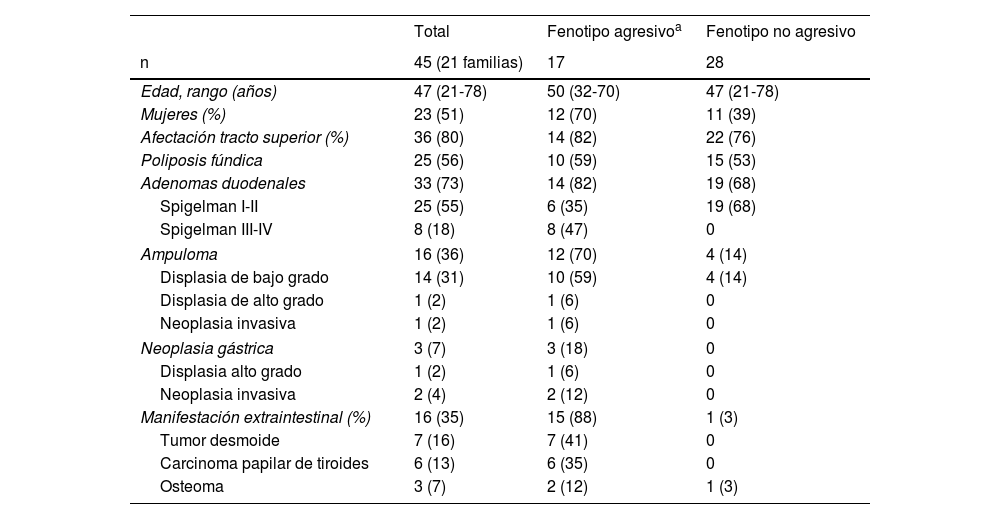
ResultadosCaracterísticas fenotípicasEn nuestro estudio registramos 45 pacientes diagnosticados de PAF, controlados en la consulta monográfica de nuestro centro, pertenecientes a 21 familias diferentes. El 88% de los pacientes presentaban manifestaciones extracolónicas (40/45). La edad media de los pacientes fue de 47años (rango 21-78años), y 23 de ellos fueron mujeres (51%). Todos los pacientes incluidos en el estudio correspondían a la variante de PAF clásica.
Los pacientes con PAF presentaron una mediana de 2 (rango 0-6) manifestaciones fenotípicas por paciente. El 38% de los pacientes (n=17) presentaron un comportamiento agresivo.
La afectación del tracto digestivo superior se observó en 36 pacientes (80%), siendo los adenomas duodenales (n=33 [73%]) y la poliposis fúndica (n=25 [57%]) los hallazgos más frecuentes. Los pacientes con adenomas duodenales fueron estratificados según la clasificación de Spigelman (I-IV): I (n=11 [33%]), II (n=14 [42%]), III (n=8 [24%]) y IV (n=0). Se detectó la presencia de ampuloma o tumor papilar, diámetro medio de 5,5mm (rango 2-15mm) en 16 de los 45 pacientes (36%), siendo los resultados del estudio histológico: displasia de bajo grado (n=14 [31%]), displasia de alto grado (n=1 [2,2%]) y neoplasia invasiva (n=1 [2,2%]). Las lesiones neoplásicas gástricas registradas fueron adenoma con displasia de alto grado (n=1) y adenocarcinoma infiltrante (n=2).
La manifestación extraintestinal más frecuente fue el tumor desmoide, presente en 7 de los 45 pacientes (16%), seguida de los carcinomas papilares de tiroides (n=6 [13%]) y de los osteomas (n=3 [6,6%]) (tabla 1).
Características demográficas y clínicas de los pacientes
| Total | Fenotipo agresivoa | Fenotipo no agresivo | |
|---|---|---|---|
| n | 45 (21 familias) | 17 | 28 |
| Edad, rango (años) | 47 (21-78) | 50 (32-70) | 47 (21-78) |
| Mujeres (%) | 23 (51) | 12 (70) | 11 (39) |
| Afectación tracto superior (%) | 36 (80) | 14 (82) | 22 (76) |
| Poliposis fúndica | 25 (56) | 10 (59) | 15 (53) |
| Adenomas duodenales | 33 (73) | 14 (82) | 19 (68) |
| Spigelman I-II | 25 (55) | 6 (35) | 19 (68) |
| Spigelman III-IV | 8 (18) | 8 (47) | 0 |
| Ampuloma | 16 (36) | 12 (70) | 4 (14) |
| Displasia de bajo grado | 14 (31) | 10 (59) | 4 (14) |
| Displasia de alto grado | 1 (2) | 1 (6) | 0 |
| Neoplasia invasiva | 1 (2) | 1 (6) | 0 |
| Neoplasia gástrica | 3 (7) | 3 (18) | 0 |
| Displasia alto grado | 1 (2) | 1 (6) | 0 |
| Neoplasia invasiva | 2 (4) | 2 (12) | 0 |
| Manifestación extraintestinal (%) | 16 (35) | 15 (88) | 1 (3) |
| Tumor desmoide | 7 (16) | 7 (41) | 0 |
| Carcinoma papilar de tiroides | 6 (13) | 6 (35) | 0 |
| Osteoma | 3 (7) | 2 (12) | 1 (3) |
Todos los pacientes de nuestro estudio presentaron variantes patogénicas en el gen APC. De los 45 pacientes incluidos, disponemos de análisis genético en 43 (95,5%). Las variantes detectadas en el estudio genético fueron: frameshift (n=24 [56%]), nonsense (n=11 [26%]), splicing (n=7 [16%]) y rearrangement (n=1).
La mayoría de variantes presentes en nuestros pacientes (80% [36/45]) suponen un codón de parada inmediato que generan una proteína truncada, y que se corresponde a las variantes tipo frameshift, nonsense y rearrangement.
Al analizar la localización de las variantes patogénicas en función del codón afectado, se observó que principalmente se localizan en el exón15 (fragmentoE) (39,1%), intrón (15,2%) y exón 15 (fragmentoG) (10,9%) (tablas 2 y 3). En la figura 1 se puede observar como la mayoría de las variantes patogénicas se localizan en el exón15, que es el de mayor tamaño del gen APC.
Análisis de las alteraciones genéticas en función del tipo y localización de los pacientes con poliposis adenomatosa familiar
| Localización | Frameshift(n = 24) | Nonsense(n = 11) | Splicing(n = 7) |
|---|---|---|---|
| Exón 5 | − | c.637C>T, p.Arg213* (1) | − |
| Exón 9 | c.1287delA, p.Gly430Afs*24 (1) | c.994C>T, p.Arg332* (1) | − |
| Exón 13 | − | c.1660C>T, p.Arg554 (3) | − |
| Intrón 13 | − | - | c.1744-2A>G (7) |
| Exón 15 B | − | c.2413C>T, p.Arg805* (3) | − |
| Exón 15 C | − | c.2795C>A, p.Ser932* (2)c.2805C>A, p.Tyr935* (1) | − |
| Exón 15 E | c.3126_3139delTCCTTCACAGAATG, p.Ser1042Argfs*13 (1)c.3183_3187delACAAA, Gln1062* (7)c.3185_3186delAA, p.Ser1063* (4)c.3186_3187delAA, p.Ser1063* (2).3204_3205delAA, p.Arg1069fs*11 (2)c.3441_3442insA, p.Ser1148Ilefs*2 (2) | - | − |
| Exón 15 G | c.3927_3931delAAAGA, p.Glu1309Aspfs*4 (4)c.4024delT, p.L1342Yfs*73 (1) | − | − |
Localización en el gen APC de las variantes patogénicas frameshift y nonsense
| Variante patogénica | Codón de terminación prematuro en la posición |
|---|---|
| c.637C>T, p.Arg213* | Codón 213 |
| c.994C>T, p.Arg332* | Codón 332 |
| c.1287delA, p.Gly430Afs*24 | Codón 454 |
| c.1660C>T, p.Arg554 | Codón 554 |
| c.2413C>T, p.Arg805* | Codón 805 |
| c.2795C>A, p.Ser932* | Codón 932 |
| c.2805C>A, p.Tyr935* | Codón 935 |
| c.3126_3139delTCCTTCACAGAATG, p.Ser1042Argfs*13 | Codón 1054 |
| c.3183_3187delACAAA, Gln1062* | Codón 1062 |
| c.3185_3186delAA, p.Ser1063* | Codón 1063 |
| c.3186_3187delAA, p.Ser1063* | Codón 1063 |
| c.3204_3205delAA, p.Arg1069fs*11 | Codón 1080 |
| c.3441_3442insA, p.Ser1148Ilefs*2 | Codón 1150 |
| c.3927_3931delAAAGA, p.Glu1309Aspfs*4 | Codón 1313 |
| c.4024delT, p.L1342Yfs*73 | Codón 1415 |

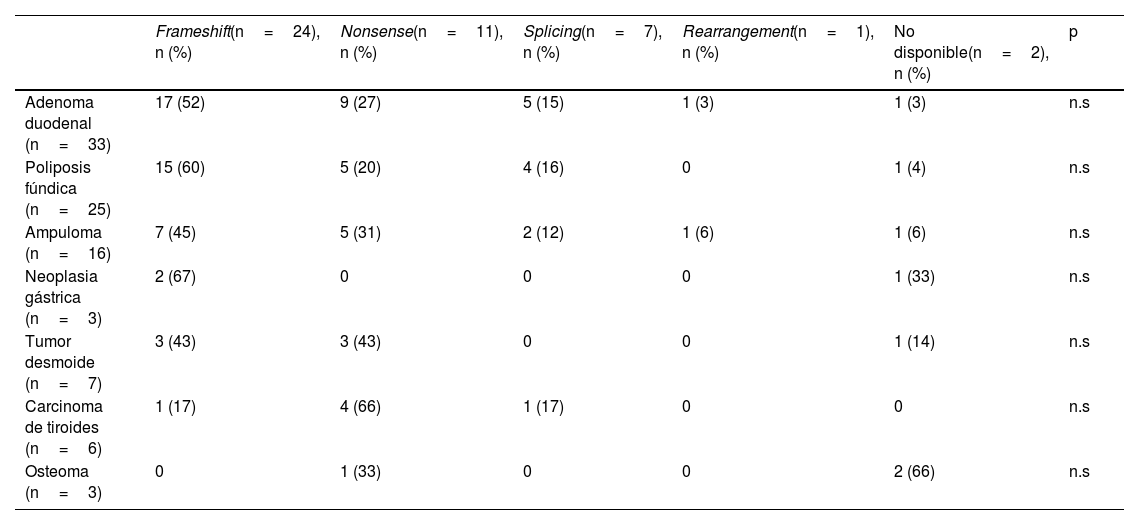
La correlación entre las diferentes manifestaciones extracolónicas con el tipo de alteración genética fue muy heterogénea, sin evidencia de diferencias significativas entre el tipo de variante con el patrón fenotípico (tabla 4). Los pacientes con variantes tipo frameshift y nonsense presentaron un mayor riesgo de desarrollar manifestaciones extracolónicas. Las variantes frameshift fueron las más prevalentes en los pacientes con manifestaciones digestivas extracolónicas, mientras que las nonsense, lo fueron en los pacientes con manifestaciones extradigestivas.
Prevalencia de las diferentes manifestaciones extracolónicas en función del tipo de variante patogénica, n (%)
| Frameshift(n=24), n (%) | Nonsense(n=11), n (%) | Splicing(n=7), n (%) | Rearrangement(n=1), n (%) | No disponible(n=2), n (%) | p | |
|---|---|---|---|---|---|---|
| Adenoma duodenal (n=33) | 17 (52) | 9 (27) | 5 (15) | 1 (3) | 1 (3) | n.s |
| Poliposis fúndica (n=25) | 15 (60) | 5 (20) | 4 (16) | 0 | 1 (4) | n.s |
| Ampuloma (n=16) | 7 (45) | 5 (31) | 2 (12) | 1 (6) | 1 (6) | n.s |
| Neoplasia gástrica (n=3) | 2 (67) | 0 | 0 | 0 | 1 (33) | n.s |
| Tumor desmoide (n=7) | 3 (43) | 3 (43) | 0 | 0 | 1 (14) | n.s |
| Carcinoma de tiroides (n=6) | 1 (17) | 4 (66) | 1 (17) | 0 | 0 | n.s |
| Osteoma (n=3) | 0 | 1 (33) | 0 | 0 | 2 (66) | n.s |
Tras un análisis exhaustivo de las correlaciones genotipo-fenotipo de las variantes germinales patogénicas de la APC, no se encontró ninguna asociación significativa. En el análisis de contingencia tampoco se demostró una asociación entre la presencia de una proteína truncada con el desarrollo de fenotipo agresivo (fig. 2).
Discusión
El 88% de nuestra cohorte de pacientes con PAF presentaron manifestaciones extracolónicas, y un tercio de ellos un fenotipo agresivo, sin demostrarse una correlación entre el tipo de variante patogénica y las manifestaciones fenotípicas. Estos datos enfatizan la importancia de su cribado y del seguimiento exhaustivo con el objetivo de disminuir el riesgo de progresión neoplásica.
En nuestro estudio incluimos una serie amplia de pacientes con PAF con la variante clásica, y con una gran variabilidad de manifestaciones extracolónicas. La afectación del tracto digestivo superior fue muy prevalente, apreciándose en el 80% de nuestros pacientes, siendo los adenomas duodenales y la poliposis fúndica los hallazgos más habituales. En nuestra serie también se detectó la presencia de neoplasia invasiva, que requirió cirugía en 3 de los 45 pacientes (6,6%), siendo muy llamativo el diagnóstico de adenocarcinoma gástrico en 2 pacientes, pese al correcto seguimiento endoscópico, por lo que es crucial realizar una exhaustiva exploración endoscópica de calidad que garantice el correcto diagnóstico óptico16. En cuanto a las manifestaciones extraintestinales, las más frecuentes fueron los tumores desmoides y los carcinomas papilares de tiroides.
La alteración genética más habitual fue la frameshift, en más de la mitad de nuestros pacientes, seguida de la nonsense y de la splicing en un porcentaje muy inferior, al igual que lo descrito en la literatura17.
En nuestro estudio no encontramos una asociación estadística entre el tipo de variante patogénica y la manifestación fenotípica, aunque sí una cierta tendencia. La presencia de una variante tipo frameshift fue la más prevalente en los pacientes con neoplasia gástrica, poliposis fúndica, adenomas duodenales, ampulomas y tumores desmoides, mientras que las variantes nonsense fueron las más prevalentes en los pacientes con carcinoma papilar de tiroides y con tumores desmoides. Las correlaciones genotipo-fenotipo en los síndromes de poliposis se han evaluado en varios estudios y se ha observado una asociación general entre la posición de la variante y la manifestación clínica, aunque con algunas inconsistencias16,18-20. Los pacientes con PAF más agresiva se relacionan con variantes entre los codones 1250 y 1464, aunque, al igual que en nuestro estudio, tampoco se encontró correlación con las manifestaciones digestivas extracolónicas4,21. La inclusión de los tumores desmoides dentro de las manifestaciones fenotípicas agresivas viene motivada por su patrón de crecimiento infiltrativo y por la propensión a la recurrencia local22, al igual que la presencia de adenomas periampulares más avanzados (SpigelmannIII-IV) por mayor riesgo de aparición de adenocarcinomas16. En estudios previos se ha demostrado que el riesgo de desarrollar adenomas de duodeno es 3-4 veces superior si la variante se sitúa entre los codones 976-106721; además, los pacientes con variantes de la línea germinal después del codón 1400 tienden a tener una poliposis duodenal más grave16. En nuestra serie se realizó resección endoscópica en todos los pacientes con ampuloma, confirmándose que los pacientes con histología más agresiva (displasia de alto grado y neoplasia invasiva) presentaban la variante frameshift en el codón 1062 y 1063.
El desarrollo de osteomas suele preceder al desarrollo de las manifestaciones intestinales23, y según algunas series pueden estar presentes en el 60-80% de los pacientes con PAF, mientras que su prevalencia es del 1-2% en la población general24. Este porcentaje es mucho más pequeño en nuestra serie (3 casos [6,6%]), prevalencia más parecida a la descrita en el registro español de 2010 (2,5%)25 y asociada a variantes entre los codones 767 y 1578 en la bibliografía26.
En el caso de los tumores desmoides, originados por sobrecrecimiento del tejido fibroaponeurótico, su prevalencia se sitúa en el 12-15% de los pacientes con PAF27, al igual que en nuestra serie, con un mayor riesgo de desarrollo si la variante es posterior al exón 1399, principalmente entre los codones 1445 y 15804,28. En contraposición a lo descrito en la bibliografía, todos nuestros pacientes presentan una variante en el gen previo al codón 1313.
La incidencia de cáncer de tiroides es del 2 al 12% en los casos con PAF, asociada a variantes patogénicas germinales entre los codones 1286 y 1513 en el gen APC21, o cuando esta se localiza en el codón 106129. Los 6 casos de nuestra serie (13%) confirman la prevalencia descrita en la literatura, siendo esta muy superior a lo descrito en el registro español, con una prevalencia del 3,3%25.
Por último, destacar que este estudio presenta varias limitaciones que deben considerarse al interpretar los resultados. En primer lugar, el diseño unicéntrico puede limitar la generalización de los hallazgos a otras poblaciones. En segundo lugar, el tamaño de la muestra podría haber influido en la capacidad para detectar asociaciones estadísticas más sólidas entre las variantes patogénicas y las manifestaciones fenotípicas extracolónicas. Un tamaño muestral mayor podría ofrecer mayor potencia estadística para analizar estas correlaciones. El tipo de análisis estadístico utilizado, la presencia de posibles factores de confusión no considerados y la falta de seguimiento longitudinal serían también limitaciones para tener en cuenta. Estas limitaciones subrayan la necesidad de estudios adicionales con un diseño multicéntrico, mayor tamaño muestral y análisis estadísticos más complejos para validar y ampliar nuestros hallazgos.
En conclusión, y pese a las limitaciones de nuestro estudio, se confirma que la afectación gastroduodenal es muy prevalente en los pacientes con PAF. Un tercio de estos pacientes presentan un fenotipo agresivo, por lo que es fundamental un exhaustivo seguimiento para disminuir la morbimortalidad de esta enfermedad, independientemente del tipo de variante patogénica que sufran. Por otro lado, el tipo de variante, y no solo su localización, podría estar implicado directamente en el fenotipo de los pacientes con PAF, por lo que es importante realizar estudios multicéntricos que nos permitan extraer evidencia suficiente como para avanzar hacia un seguimiento más personalizado en función del genotipo de cada paciente. Aunque en nuestro estudio no se observó una correlación significativa entre las variantes patogénicas y las manifestaciones fenotípicas, ciertas tendencias sugieren que las variantes genéticas podrían influir en la estrategia de manejo y la vigilancia más intensiva. Un mayor conocimiento del gen APC y de su funcionamiento podría ayudar a definir con mayor precisión el seguimiento clínico de los pacientes con PAF.
Consideraciones éticasHemos seguido los protocolos de nuestro centro de trabajo sobre la publicación de datos de pacientes.
Conflicto de interesesLos autores del manuscrito no presentan ningún conflicto de intereses.

