






Los sarcomas renales primarios son sumamente raros y representan entre el 1% y el 3% de las neoplasias malignas de riñón, siendo las formas histológicas más frecuentes el leiomiosarcoma y el liposarcoma. El sarcoma pleomórfico indiferenciado renal primario es una variante rara de mal pronóstico, con alrededor de 60 casos descritos en la literatura. Presentamos el caso de una mujer de 52 años con un tumor renal derecho, tratada quirúrgicamente y cuyo diagnóstico histológico confirmado con estudio inmunohistoquímico fue sarcoma pleomórfico indiferenciado de la variedad de células gigantes.
Primary sarcomas of the kidney are a rare entity and they account for 1 - 3 % of kidney malignancies, with the most common histological types being leiomyosarcoma and liposarcoma. Primary undifferentiated pleomorphic sarcoma of the kidney is a rare variant with a poor prognosis, with about 60 cases described in the literature. The case is presented of a 52-year-old female with a right renal tumour. It was surgically treated, with an immunohistochemistry-confirmed histological diagnosis of undifferentiated pleomorphic sarcoma of the giant cell variant.
El sarcoma pleomórfico indiferenciado (SPI), antiguamente denominado fibrohistiocitoma maligno pleomórfico, es la neoplasia maligna de partes blandas más común en la vida adulta1–3. Fue descrito por primera vez por O’Brien y Stout en 1964, siendo los lugares más afectados las extremidades (67-75%) y el retroperitoneo (6-16%)4–6. Sin embargo, también puede presentarse en otras localizaciones del sistema genitourinario, como por ejemplo, en orden de frecuencia: vejiga, próstata, cordón espermático, cápsula y parénquima renal4,6,7. El SPI primario renal es extremadamente raro; su presentación clínica y manejo quirúrgico no difieren de las del carcinoma renal convencional, y para su diagnóstico se requiere de técnicas de inmunohistoquímica que lo confirmen7–9. Presentamos el caso de una mujer de 52 años con SPI renal primario, tratada quirúrgicamente, y que evolucionó desfavorablemente.
Presentación del casoPaciente mujer de 52 años, natural y procedente de Chimbote, G2P2002 con antecedente familiar directo de hepatocarcinoma. Ingresó en la institución por cursar durante los 4 meses previos con dolor en el hipocondrio y flanco derechos, pérdida ponderal de 10kg, saciedad precoz, náuseas y vómitos sin presencia de hematuria macroscópica. En el examen físico preferencial la paciente se encontró adelgazada, con masa palpable, móvil, de consistencia incrementada en el hemiabdomen derecho.
En la tomografía computarizada se observó una masa tumoral de 223×190mm, dependiente del polo superior del riñón derecho, encapsulada, con tenue trabeculado captador de contraste y amplia necrosis central (20UH). La tumoración estaba en contacto con el pedículo vascular, sin signos de infiltración, manteniendo un adecuado plano de clivaje en el borde hepático. No se observaban adenopatías retroperitoneales (fig. 1). En la radiografía de tórax no se encontraron lesiones compatibles con metástasis. Los exámenes de laboratorio de rutina mostraron parámetros normales.
.")
Con estos resultados la paciente fue sometida a nefrectomía radical derecha con adrenalectomía ipsilateral y disección de adenopatías retrocavas. En el laboratorio de patología se recibió la pieza quirúrgica, cuyo peso fue de 1,619g; la grasa perirrenal se encontraba firmemente adherida al riñón; el mismo mostraba una superficie multinodular y de color pardo violáceo. Al corte se identificó un tumor de 20.0×18.0cm que destruía los cálices renales e infiltraba la capsula renal, la grasa perirrenal y el seno renal. El tumor era de color pardo claro, con áreas de hemorragia y necrosis. También se observó uréter de 14.0×6.0cm y glándula suprarrenal de 5.0×1.8cm, sin alteraciones macroscópicas significativas. Además, se recibió un ganglio retrocavo congestivo de 3.5cm (fig. 2).

En la microscopia se observó una neoplasia maligna fusocelular de tipo sarcomatoide, pleomórfica, con células gigantes multinucleadas, alto índice mitótico (12-15 mitosis/10 CAP), necrosis tumoral y embolia vascular. La neoplasia infiltraba la cápsula renal, la grasa renal y el seno renal. El uréter, la glándula suprarrenal y el borde quirúrgico se encontraron libres de neoplasia, al igual que el ganglio retrocavo (0/1) (fig. 3A-C).
: A. 10×. Vista panorámica del tumor renal en el que se observa proliferación neoplásica fusocelular con patrón de crecimiento estoriforme. B. 40×. Microfotografía a mayor aumento, en la que se observan células fusiformes malignas en relación con sarcoma. Nótese la hipercromasia nuclear. C. 100×. Detalle a alto poder, células pleomórficas gigantes con multinucleación.")
Microscopía (H-E): A. 10×. Vista panorámica del tumor renal en el que se observa proliferación neoplásica fusocelular con patrón de crecimiento estoriforme. B. 40×. Microfotografía a mayor aumento, en la que se observan células fusiformes malignas en relación con sarcoma. Nótese la hipercromasia nuclear. C. 100×. Detalle a alto poder, células pleomórficas gigantes con multinucleación.
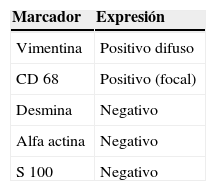
El estudio inmunohistoquímico mostró positividad difusa a vimentina y focal a CD 68, confirmando la naturaleza mesenquimal de la neoplasia; el resto de marcadores específicos a otros linajes celulares resultó negativo (tabla 1). Con estos resultados se concluyó como diagnóstico sarcoma pleomórfico indiferenciado renal primario de la variedad de células gigantes (fig. 4A-E).
. A. Vimentina: positivo intenso. B. CD 68: positivo focal. C. Desmina: negativo. D. Alfa actina: negativo. E. S 100: negativo.")
La paciente toleró el procedimiento quirúrgico y fue dada de alta el séptimo día del postoperatorio. Tres meses después reingresó en el instituto por presentar disnea de leve y mediano esfuerzo, sin tolerancia al decúbito. En la radiografía de tórax se observó una masa tumoral macronodular en la región parietocostal derecha, asociada a derrame y atelectasia compresiva. La paciente entró en una fase de deterioro progresivo y finalmente falleció por compromiso multisistémico.
DiscusiónLos sarcomas primarios de riñón son neoplasias de origen mesenquimatoso muy poco frecuentes que constituyen entre el 1% y el 3% de los tumores renales malignos1,2,5. Dependiendo de su tipo histológico se han descrito casos de leiomiosarcoma, rabdomiosarcoma, liposarcoma, angiosarcoma, sarcoma pleomórfico, sarcoma sinovial y osteosarcoma4,7,10–12. El más común de estos tumores es el leiomiosarcoma, que representa entre el 40% y el 60% de los casos publicados2,3.
El SPI, antes llamado fibrohistiocitoma pleomórfico maligno5,13, es una variante inusual, con alrededor de 60 casos publicados en la literatura mundial, la mayoría del siglo pasado; no se encontraron reportes nacionales en la revisión bibliográfica1,7. No existen criterios definidos para el diagnóstico del sarcoma renal primario; sin embargo, se sugieren los siguientes: 1) ausencia de antecedentes de sarcoma en otra localización anatómica (excepto por la presencia de metástasis); 2) un estudio macroscópico que demuestre el origen renal de tumor; 3) exclusión del diagnóstico de carcinoma renal de patrón sarcomatoide; y 4) de existir una metástasis esta debe ser más pequeña que el tumor renal2,13,14. Su origen sigue siendo desconocido y la hipótesis más aceptada es la que considera las células mesenquimatosas pluripotenciales como sus precursoras1,6,9. Otra teoría aceptada es la que involucra las células del sistema fagocítico mononuclear15–17.
El SPI primario de riñón ocurre principalmente en adultos entre la quinta y séptima décadas de la vida, y compromete a ambos sexos por igual1,16,18,. La clínica es inespecífica, y por lo general depende del tamaño de la masa tumoral. El síntoma inicial es el dolor en el flanco comprometido asociado a pérdida de peso, como se describe en el caso que presentamos; también se reporta fiebre, masa palpable y más raramente hematuria15,18,19.
Los estudios de imagen permiten documentar la composición, densidad, extensión y la relación con órganos y estructuras adyacentes15. La radiografía abdominal presenta aumento de densidad con efecto de masa a nivel del flanco comprometido, y la ecografía permite identificar la localización, el tamaño y la estructura renal interna15. Aunque el SPI es indistinguible tanto clínica como radiográficamente del carcinoma renal, utilizando imágenes de mayor resolución, como la tomografía computarizada y la resonancia magnética se puede orientar el diagnóstico14,15,20. Se han descrito características radiológicas sugestivas de SPI: 1) tumores con varias intensidades de señales en la resonancia magnética; 2) tumores hipovasculares o avasculares en la angiografía; (3) tomográficamente presencia de áreas hipodensas (degeneración quística) e hiperdensas (calcificaciones); y 4) tumores mayores de 10cm que no invaden la vena renal ni la cava; estos últimos criterios se encontraban presentes en nuestro caso6,15.
El diagnóstico definitivo es histológico con confirmación inmunohistoquímica6,18. El riñón más afectado suele ser el izquierdo1, a diferencia de nuestro caso, en el que se trató del derecho. Los SPI renales son de gran tamaño y presentan características similares a los sarcomas de partes blandas; son multilobulados, de color amarillo grisáceo, deficientemente encapsulados y con extensas áreas de hemorragia y necrosis7,13. Microscópicamente se caracterizan por presentar células fusiformes muy pleomórficas y multinucleadas que se entremezclan con fibroblastos y macrófagos13,18. La actividad mitótica es intensa, identificándose infiltrado inflamatorio y áreas de necrosis15,16. Se han descrito 5 variantes histológicas, las cuales en orden de frecuencia son: estoriforme-pleomórfico, mixoide, de células gigantes, inflamatorio y angiomatoso1,6,13. Desde el punto de vista inmunohistoquímico, se establece el diagnóstico por exclusión, utilizando el panel de marcadores clásico para sarcomas de partes blandas5. Lo más importante es la negatividad de las reacciones a linajes celulares específicos y la positividad intensa a vimentina y variable a CD 68, quimotripsina, Leu-M3 y otros marcadores histiocíticos7,15,18. El estudio histológico del caso que reportamos presenta características morfológicas de sarcoma de alto grado, en el que predominan las células gigantes multinucleadas. La positividad a vimentina y CD 68 confirma la naturaleza mesenquimatosa del tumor, siendo negativos los otros marcadores utilizados para determinar el linaje específico.
El diagnóstico diferencial incluye lesiones benignas, como la pielonefritis xantogranulomatosa. y malignas como el carcinoma sarcomatoide renal y otros tipos de sarcoma6,13,14. En el caso específico de la variedad células gigantes debe distinguirse del leiomiosarcoma de células gigantes osteoclásticas, del carcinoma indiferenciado de tipo osteoclastoma y del carcinoma indiferenciado del tipo de células gigantes, así como de enfermedades muy poco frecuentes de riñón que tienen morfología e inmunofenotipo propios16. Se han identificado como factores asociados a mal pronóstico la edad avanzada, el tamaño de la masa tumoral, la profundidad de infiltración, la necrosis tumoral, un alto índice mitótico y las metástasis a distancia17,18. El componente inflamatorio y el grado histológico también han sido incluidos1,19. En el reporte que presentamos identificamos la mayoría de los factores descritos.
El SPI es un tumor agresivo y de mal pronóstico en el que la cirugía radical constituye la primera línea de tratamiento con intención curativa14,16,17,20. La recurrencia local es alta, de entre el 50% y el 82%, en un rango temporal de 3 a 24 meses tras la cirugía7,15,18. Está asociado a metástasis pulmonar en un 82% y ganglionar en un 32%1, y con menos frecuencia también se ven afectados el hígado, el hueso y la médula ósea15. La utilidad de la quimioterapia y la radioterapia no está definida debido al escaso número de casos reportados14,16; sin embargo, en algunos artículos, la quimioterapia ha demostrado un ligero beneficio en la supervivencia subsecuente al tratamiento quirúrgico (10 a 18 meses libres de enfermedad)18,20. En contraste, la radioterapia adyuvante no ha demostrado resultados beneficiosos18. La mortalidad es alta, aproximadamente del 66% a 2 años tras la nefrectomía radical1.
ConclusiónEl SPI renal primario de la variante de células gigantes es una neoplasia rara, extremadamente agresiva y de mal pronóstico, cuyo diagnóstico preoperatorio es sumamente difícil. Para confirmarlo se requiere de estudio histológico e inmunohistoquímico. La cirugía es el único tratamiento con intención curativa, en tanto que la terapia adyuvante no ha demostrado beneficios.
FinanciaciónLos autores no recibieron patrocinio alguno para este trabajo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.