

Revisar la prevalencia de resistencia a la aspirina en pacientes de alto riesgo cardiovascular y, de forma secundaria, investigar la epidemiología, los mecanismos de acción de este fenómeno y las consecuencias clínicas que se derivan de ello.
Material y métodosSe realizó una búsqueda en PubMed, EMBASE y Reviews Database de artículos publicados en inglés y en español hasta noviembre de 2008 referidos a resistencia a la aspirina. Se hizo también un seguimiento de las referencias para recuperar aquéllas consideradas relevantes para los objetivos secundarios de esta revisión.
ResultadosLa resistencia a la aspirina se describe entre un 0 y un 57%, lo que se traduce en una disminución de la eficacia protectora frente a eventos cerebrovasculares y cardiovasculares. Numerosos factores modificables, así como otros no modificables, influyen en la eficacia del efecto antiagregante. Las estrategias por seguir para contrarrestar esta antiagregación disminuida pueden dirigirse a un aumento de dosis de aspirina o a la terapia dual junto con otros agentes antiplaquetarios.
ConclusionesLa falta de respuesta a la aspirina disminuye su eficacia protectora. Sin embargo, la falta de una definición de resistencia a la aspirina universalmente consensuada y la inexistencia de un método diagnóstico de referencia que identifique con fiabilidad a pacientes resistentes, así como los diferentes mecanismos de acción implicados en la agregación plaquetaria, cuestionan la relevancia clínica de este fenómeno. Son necesarios más estudios clínicos adecuadamente diseñados para detectar pacientes con verdadera resistencia a la aspirina y así conseguir prevenir de manera más eficaz la morbimortalidad cardiovascular.
The purpose of this study is to review the prevalence of aspirin resistance in patients with a high risk of cardiovascular events, and secondly, to investigate its epidemiology and mechanism of action, and the clinical consequences it can provoke.
Material and methodsA search was run on PubMed, EMBASE and Reviews Database for English or Spanish articles on aspirin resistance published up to November 2008. Additional studies were obtained by searching the reference lists in the selected articles for articles relevant to our secondary objectives.
ResultsAspirin resistance is described as affecting 0 to 57% of the population, and is related to a decreased protective effect against strokes and cardiovascular events. Many modifiable and unmodifiable factors can affect the efficacy of antiplatelet drugs. Possible strategies for overcoming this decreased antiaggregant effect include increasing the aspirin dosage or dual therapy with another antiplatelet agent.
ConclusionsLack of response to aspirin decreases its protective effects. However, lack of a standard definition for aspirin resistance, the absence of diagnostic reference methods to identify resistant patients, and the different mechanisms of action involved in platelet aggregation call the clinical importance of this fact into question. Additional well-designed studies are needed to detect patients with real resistance in order to have more effective prevention of cardiovascular morbidity and mortality.
Las enfermedades cardiovasculares y cerebrovasculares están entre las principales causas de morbimortalidad en los países desarrollados. La aspirina o ácido acetilsalicílico (AAS) es el tratamiento antiagregante plaquetario más utilizado para la profilaxis de eventos tromboembólicos. En pacientes con alto riesgo de enfermedad cardiovascular la terapia con AAS ha demostrado reducir el riesgo de infarto agudo de miocardio (IAM) no mortal, ictus no mortal o muerte por causas vasculares1,2.
El efecto antiagregante del AAS se consigue a través de la inhibición permanente de la enzima prostaglandina (PG) sintetasa o ciclooxigenasa (COX)3. La enzima COX se presenta en 2 isoformas: por una parte, la COX-1, que es una enzima constitutiva presente en la mayoría de las células y, por otra parte, la COX-2, que se expresa únicamente en respuesta a estímulos inflamatorios. La inhibición irreversible de la COX-1 bloquea la conversión del ácido araquidónico (AA) a PG, como la PGH2, precursora del tromboxano A2 (TXA2)3. Este mediador, junto a otros mediadores biológicos como la adenosinadifosfato (ADP) o el fibrinógeno, actúa favoreciendo un estado protrombótico.
El AAS es 170 veces más potente en inhibir la enzima COX-1 que la enzima COX-2, por lo que en dosis bajas (75–300mg) sólo se inhibe la enzima COX-1 (efecto antiagregante). A pesar de que el AAS tiene una corta semivida (aproximadamente 30min), como inactiva irreversiblemente la enzima COX-1 de plaquetas y únicamente un 10% de las plaquetas son renovadas diariamente, una única dosis diaria es suficiente para inhibir el 90% de la producción de TXA2.
Sin embargo, se han observado casos en los que el tratamiento antiagregante es subóptimo, por lo que determinados pacientes experimentan eventos cardiovasculares a pesar de la administración correcta del AAS4. Esto se denomina “resistencia o falta de respuesta al AAS”. Este término, aunque no tiene una definición aceptada universalmente, se puede orientar desde dos enfoques distintos. Desde el punto de vista clínico, la resistencia (o no respuesta) al AAS se define como la aparición de eventos cardiovasculares tromboembólicos a pesar del tratamiento continuado con AAS en dosis terapéuticas. Pero, debido a la naturaleza multifactorial de los procesos aterotrombóticos5, no es preciso hablar exclusivamente de resistencia al AAS en este contexto. Por tanto, se suele emplear más frecuentemente la definición bioquímica, en la que resistencia se define bien como fracaso del AAS, en dosis terapéuticas, en prolongar el tiempo de sangrado, que es una medida primaria de la función plaquetaria, o bien como fracaso en la reducción de la producción de TXA26.
Los métodos más empleados que se utilizan para medir el grado de inhibición plaquetaria se describen a continuación7:
- •
Test in vivo:
- ○
Concentración de tromboxano B2 urinario: el metabolito estable del TXA2, el 11-dehidrotromboxano B2, es medido en la orina como marcador de la activación plaquetaria y, por tanto, de la respuesta a fármacos antitrombóticos.
- ○
Tiempo de sangrado: se mide cuánto dura la hemorragia a consecuencia de una punción practicada sobre la piel. Se trata de una técnica muy poco empleada porque es escasamente reproducible.
- ○
- •
Test in vitro. Se han desarrollado múltiples técnicas de laboratorio para medir la activación plaquetaria. Sin embargo, hoy en día ninguna es considerada de referencia, ya que no valoran la agregación plaquetaria en su conjunto, puesto que existen diversas vías de activación de las plaquetas. Sin embargo, son métodos mucho más empleados que los test in vivo.
- ○
Agregometría óptica: se trata de una técnica espectrofotométrica que mide la transmisión de la luz a través de una muestra de plasma rico en plaquetas. La agregación se induce con un determinado agonista plaquetario (AA, ADP, epinefrina, etc.) en función del fármaco antiagregante que se valore y teniendo en cuenta el distinto mecanismo de acción entre ellos.
- ○
Verify Now (RPFA [Ultegra-Rapid Platelet Function Assay]): turbidímetro que se basa en el mismo principio que el método anterior con la ventaja de ser más rápido y de requerir un pequeño volumen de muestra. El resultado se expresa en unidades arbitrarias de respuesta al agonista.
- ○
PFA-100 (Platelet Function Analyzer): analizador que valora automáticamente la agregación plaquetaria inducida por una alta velocidad de flujo y por una mezcla de activadores plaquetarios, colágeno/epinefrina o colágeno/ADP que están adheridos a una membrana capilar. Las plaquetas interactúan con la membrana y se ocluye la apertura. La respuesta hemostática se expresa como el tiempo necesario para obstruir el flujo en el interior de un capilar, el cual será de menor a mayor agregación plaquetaria.
- ○
El objetivo primario de esta revisión narrativa es revisar la prevalencia de resistencia al AAS en pacientes de alto riesgo cardiovascular en tratamiento con este antiagregante para la prevención secundaria de eventos cardiovasculares y cerebrovasculares. Secundariamente, se hace una descripción de los posibles mecanismos de acción que explican este fenómeno, las consecuencias clínicas que se derivan de ello y las posibles estrategias para mejorar la respuesta antigregante en estos pacientes.
Material y métodosFuentes de información: estrategia de búsqueda.
Se hizo una búsqueda bibliográfica en PubMed de todos los artículos publicados hasta noviembre de 2008, introduciendo en el MESH las palabras clave “aspirin” AND “drug resistance”. Se establecieron como límites de búsqueda publicaciones en humanos en inglés y en español. Tras recuperar un alto número de artículos se limitó la búsqueda a través de dos estrategias distintas, por una parte se limitó a “clinical trial”, “meta-analysis” y “randomized controlled trial”, y por la otra, se limitó a “review” y “practice guidelines”. Adicionalmente, se realizó otra búsqueda bibliográfica en EMBASE, combinando los términos “aspirin” AND “drug resistance” OR “disease resistance” OR “resistance blood vessel” OR “therapy resistance”.
Otras bases de datos revisadas fueron Reviews database—Cochrane Database of Systematic Reviews, American College of Physicians Journal Club, Database of Abstracts of Reviews of Effects y Cochrane Central Register of Controlled Trials.
Posteriormente, se realizó una búsqueda manual de los artículos de interés citados en la bibliografía de los artículos anteriormente seleccionados.
De los artículos seleccionados, se excluyeron de la revisión para hallar la prevalencia aquellos que no cumplían los siguientes criterios de inclusión:
- •
Pacientes mayores de 18 años en tratamiento con AAS para prevención secundaria de eventos cardiovasculares o cerebrovasculares.
- •
Definición de resistencia al AAS.
- •
Definición de los métodos de medida de función plaquetaria.
Muchos de los artículos excluidos se tuvieron en cuenta para la descripción de los objetivos secundarios de la presente revisión.
ResultadosEn total se recuperaron de Pubmed y EMBASE 190 referencias, de las que se seleccionaron cuarenta y tres según el resumen y el título. De las otras bases de datos revisadas, eliminando los duplicados, aparecieron 110 referencias nuevas, de las que se seleccionaron sólo dos por su título y por su resumen. En la búsqueda manual posterior se añadieron 2 artículos en húngaro y en polaco, de los que sólo se obtuvo el resumen.
Para realizar la revisión de la prevalencia, se seleccionaron 33 artículos que cumplían los criterios de inclusión de los 47 artículos recuperados inicialmente.
Prevalencia de la resistenciaSe revisó la prevalencia de resistencia al AAS en pacientes en tratamiento antiagregante con diferentes situaciones clínicas en los ensayos clínicos seleccionados. La gran mayoría de estos estudios incluyeron al menos un test in vitro para determinar la agregación plaquetaria.
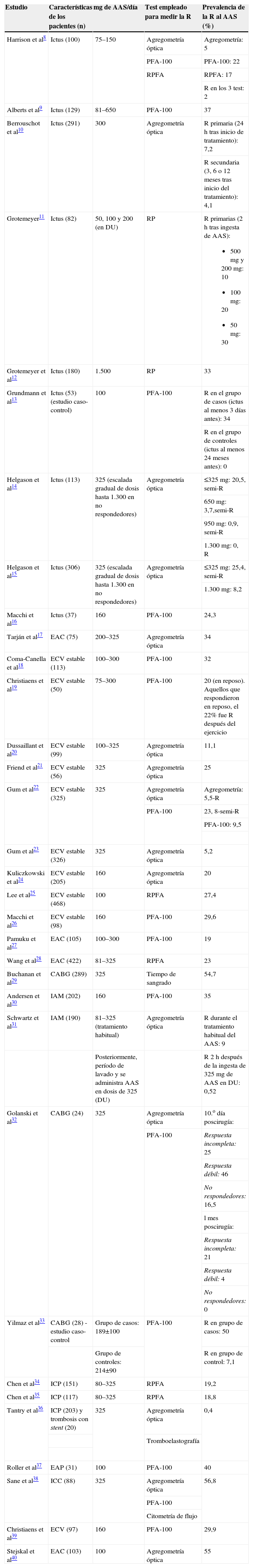
La prevalencia hallada muestra un rango de variación muy amplio, entre un 0,4–57%8–40 (tabla 1).
Prevalencia de resistencia al ácido acetilsalicílico en prevención secundaria
| Estudio | Características de los pacientes (n) | mg de AAS/día | Test empleado para medir la R | Prevalencia de la R al AAS (%) |
| Harrison et al8 | Ictus (100) | 75–150 | Agregometría óptica | Agregometría: 5 |
| PFA-100 | PFA-100: 22 | |||
| RPFA | RPFA: 17 | |||
| R en los 3 test: 2 | ||||
| Alberts et al9 | Ictus (129) | 81–650 | PFA-100 | 37 |
| Berrouschot et al10 | Ictus (291) | 300 | Agregometría óptica | R primaria (24h tras inicio de tratamiento): 7,2 |
| R secundaria (3, 6 o 12 meses tras inicio del tratamiento): 4,1 | ||||
| Grotemeyer11 | Ictus (82) | 50, 100 y 200 (en DU) | RP | R primarias (2h tras ingesta de AAS):
|
| Grotemeyer et al12 | Ictus (180) | 1.500 | RP | 33 |
| Grundmann et al13 | Ictus (53) (estudio caso-control) | 100 | PFA-100 | R en el grupo de casos (ictus al menos 3 días antes): 34 |
| R en el grupo de controles (ictus al menos 24 meses antes): 0 | ||||
| Helgason et al14 | Ictus (113) | 325 (escalada gradual de dosis hasta 1.300 en no respondedores) | Agregometría óptica | ≤325mg: 20,5, semi-R |
| 650mg: 3,7,semi-R | ||||
| 950mg: 0,9, semi-R | ||||
| 1.300mg: 0, R | ||||
| Helgason et al15 | Ictus (306) | 325 (escalada gradual de dosis hasta 1.300 en no respondedores) | Agregometría óptica | ≤325mg: 25,4, semi-R |
| 1.300mg: 8,2 | ||||
| Macchi et al16 | Ictus (37) | 160 | PFA-100 | 24,3 |
| Tarján et al17 | EAC (75) | 200–325 | Agregometría óptica | 34 |
| Coma-Canella et al18 | ECV estable (113) | 100–300 | PFA-100 | 32 |
| Christiaens et al19 | ECV estable (50) | 75–300 | PFA-100 | 20 (en reposo). Aquellos que respondieron en reposo, el 22% fue R después del ejercicio |
| Dussaillant et al20 | ECV estable (99) | 100–325 | Agregometría óptica | 11,1 |
| Friend et al21 | ECV estable (56) | 325 | Agregometría óptica | 25 |
| Gum et al22 | ECV estable (325) | 325 | Agregometría óptica | Agregometría: 5,5-R |
| PFA-100 | 23, 8-semi-R | |||
| PFA-100: 9,5 | ||||
| Gum et al23 | ECV estable (326) | 325 | Agregometría óptica | 5,2 |
| Kuliczkowski et al24 | ECV estable (205) | 160 | Agregometría óptica | 20 |
| Lee et al25 | ECV estable (468) | 100 | RPFA | 27,4 |
| Macchi et al26 | ECV estable (98) | 160 | PFA-100 | 29,6 |
| Pamuku et al27 | EAC (105) | 100–300 | PFA-100 | 19 |
| Wang et al28 | EAC (422) | 81–325 | RPFA | 23 |
| Buchanan et al29 | CABG (289) | 325 | Tiempo de sangrado | 54,7 |
| Andersen et al30 | IAM (202) | 160 | PFA-100 | 35 |
| Schwartz et al31 | IAM (190) | 81–325 (tratamiento habitual) | Agregometría óptica | R durante el tratamiento habitual del AAS: 9 |
| Posteriormente, período de lavado y se administra AAS en dosis de 325 (DU) | R 2h después de la ingesta de 325mg de AAS en DU: 0,52 | |||
| Golanski et al32 | CABG (24) | 325 | Agregometría óptica | 10.o día poscirugía: |
| PFA-100 | Respuesta incompleta: 25 | |||
| Respuesta débil: 46 | ||||
| No respondedores: 16,5 | ||||
| l mes poscirugía: | ||||
| Respuesta incompleta: 21 | ||||
| Respuesta débil: 4 | ||||
| No respondedores: 0 | ||||
| Yilmaz et al33 | CABG (28) -estudio caso-control | Grupo de casos: 189±100 | PFA-100 | R en grupo de casos: 50 |
| Grupo de controles: 214±90 | R en grupo de control: 7,1 | |||
| Chen et al34 | ICP (151) | 80–325 | RPFA | 19,2 |
| Chen et al35 | ICP (117) | 80–325 | RPFA | 18,8 |
| Tantry et al36 | ICP (203) y trombosis con stent (20) | 325 | Agregometría óptica | 0,4 |
| Tromboelastografía | ||||
| Roller et al37 | EAP (31) | 100 | PFA-100 | 40 |
| Sane et al38 | ICC (88) | 325 | Agregometría óptica | 56,8 |
| PFA-100 | ||||
| Citometría de flujo | ||||
| Christiaens et al39 | ECV (97) | 160 | PFA-100 | 29,9 |
| Stejskal et al40 | EAC (103) | 100 | Agregometría óptica | 55 |
AAS: ácido acetilsalicílico; CABG: injerto de by-pass arteriocoronario; DU: dosis única; EAC: enfermedad arterial coronaria; EAP: enfermedad arterial periférica; ECV: enfermedad cardiovascular; IAM: infarto agudo de miocardio; ICC: insuficiencia cardíaca congestiva; ICP: intervención coronaria percutánea; PFA: Platelet Function Analyzer; R: resistencia; RP: ratio de agregación plaquetaria; semi-R: semirresistencia.
En el metaanálisis de Hovens et al41 se establece una prevalencia media de resistencia al AAS de un 24% (IC del 95%: 20–28), es decir, uno de cada cuatro pacientes es susceptible de ser resistente al tratamiento. Un resultado similar obtuvieron Crescente et al42, quienes determinaron una prevalencia del 27% cuando la agregación plaquetaria fue medida por PFA-100.
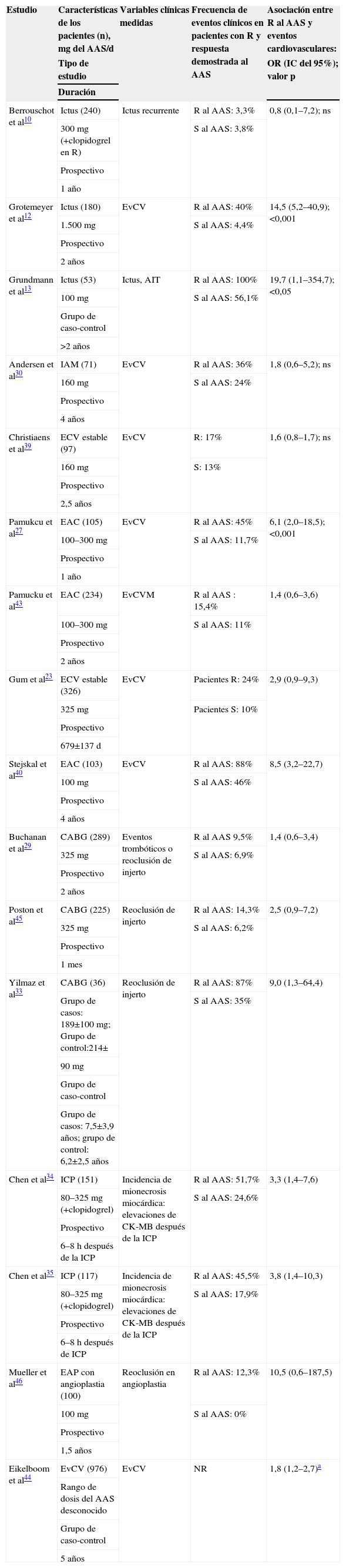
Asociación de resistencia al ácido acetilsalicílico con eventos protrombóticosLa relevancia clínica de este fenómeno se ha investigado en pacientes con antecedentes de enfermedad cardiovascular estable23,39, enfermedad arterial coronaria13,27,30,40,43,44, accidentes cerebrovasculares10,12,13,44, by-pass arteriocoronario29,33,45, enfermedad vascular periférica46 e intervenciones coronarias percutáneas34,35 (tabla 2).
Asociación entre resistencia al ácido acetilsalicílico y aparición de eventos cardiovasculares
| Estudio | Características de los pacientes (n), mg del AAS/d | Variables clínicas medidas | Frecuencia de eventos clínicos en pacientes con R y respuesta demostrada al AAS | Asociación entre R al AAS y eventos cardiovasculares: |
| Tipo de estudio | OR (IC del 95%); valor p | |||
| Duración | ||||
| Berrouschot et al10 | Ictus (240) | Ictus recurrente | R al AAS: 3,3% | 0,8 (0,1–7,2); ns |
| 300mg (+clopidogrel en R) | S al AAS: 3,8% | |||
| Prospectivo | ||||
| 1 año | ||||
| Grotemeyer et al12 | Ictus (180) | EvCV | R al AAS: 40% | 14,5 (5,2–40,9);<0,001 |
| 1.500mg | S al AAS: 4,4% | |||
| Prospectivo | ||||
| 2 años | ||||
| Grundmann et al13 | Ictus (53) | Ictus, AIT | R al AAS: 100% | 19,7 (1,1–354,7);<0,05 |
| 100mg | S al AAS: 56,1% | |||
| Grupo de caso-control | ||||
| >2 años | ||||
| Andersen et al30 | IAM (71) | EvCV | R al AAS: 36% | 1,8 (0,6–5,2); ns |
| 160mg | S al AAS: 24% | |||
| Prospectivo | ||||
| 4 años | ||||
| Christiaens et al39 | ECV estable (97) | EvCV | R: 17% | 1,6 (0,8–1,7); ns |
| 160mg | S: 13% | |||
| Prospectivo | ||||
| 2,5 años | ||||
| Pamukcu et al27 | EAC (105) | EvCV | R al AAS: 45% | 6,1 (2,0–18,5);<0,001 |
| 100–300mg | S al AAS: 11,7% | |||
| Prospectivo | ||||
| 1 año | ||||
| Pamucku et al43 | EAC (234) | EvCVM | R al AAS : 15,4% | 1,4 (0,6–3,6) |
| 100–300mg | S al AAS: 11% | |||
| Prospectivo | ||||
| 2 años | ||||
| Gum et al23 | ECV estable (326) | EvCV | Pacientes R: 24% | 2,9 (0,9–9,3) |
| 325mg | Pacientes S: 10% | |||
| Prospectivo | ||||
| 679±137d | ||||
| Stejskal et al40 | EAC (103) | EvCV | R al AAS: 88% | 8,5 (3,2–22,7) |
| 100mg | S al AAS: 46% | |||
| Prospectivo | ||||
| 4 años | ||||
| Buchanan et al29 | CABG (289) | Eventos trombóticos o reoclusión de injerto | R al AAS 9,5% | 1,4 (0,6–3,4) |
| 325mg | S al AAS: 6,9% | |||
| Prospectivo | ||||
| 2 años | ||||
| Poston et al45 | CABG (225) | Reoclusión de injerto | R al AAS: 14,3% | 2,5 (0,9–7,2) |
| 325mg | S al AAS: 6,2% | |||
| Prospectivo | ||||
| 1 mes | ||||
| Yilmaz et al33 | CABG (36) | Reoclusión de injerto | R al AAS: 87% | 9,0 (1,3–64,4) |
| Grupo de casos: 189±100mg; Grupo de control:214± | S al AAS: 35% | |||
| 90mg | ||||
| Grupo de caso-control | ||||
| Grupo de casos: 7,5±3,9 años; grupo de control: 6,2±2,5 años | ||||
| Chen et al34 | ICP (151) | Incidencia de mionecrosis miocárdica: elevaciones de CK-MB después de la ICP | R al AAS: 51,7% | 3,3 (1,4–7,6) |
| 80–325mg (+clopidogrel) | S al AAS: 24,6% | |||
| Prospectivo | ||||
| 6–8h después de la ICP | ||||
| Chen et al35 | ICP (117) | Incidencia de mionecrosis miocárdica: elevaciones de CK-MB después de la ICP | R al AAS: 45,5% | 3,8 (1,4–10,3) |
| 80–325mg (+clopidogrel) | S al AAS: 17,9% | |||
| Prospectivo | ||||
| 6–8h después de ICP | ||||
| Mueller et al46 | EAP con angioplastia (100) | Reoclusión en angioplastia | R al AAS: 12,3% | 10,5 (0,6–187,5) |
| 100mg | S al AAS: 0% | |||
| Prospectivo | ||||
| 1,5 años | ||||
| Eikelboom et al44 | EvCV (976) | EvCV | NR | 1,8 (1,2–2,7)a |
| Rango de dosis del AAS desconocido | ||||
| Grupo de caso-control | ||||
| 5 años |
AAS: ácido acetilsalicílico; AIT: accidente isquémico transitorio; CABG: injerto de by-pass arteriocoronario; CK-MB: creatinin-kinasa subtipo MB; d: días; EAC: enfermedad arterial coronaria; EAP: enfermedad arterial periférica; EvCV: eventos cardiovasculares y cerebrovasculares (fatales y/o no fatales); IC: intervalo de confianza; ICP: intervención coronaria percutánea; NR: no revelado; OR: odds ratio; R: resistencia; S: sensibilidad.
La gran mayoría de estos estudios hallan una correlación significativa entre la resistencia al AAS y un mayor riesgo de eventos cardiovasculares y cerebrovasculares respecto a los pacientes sensibles a su efecto: mayor frecuencia de accidente cerebrovascular, muerte cardiovascular, IAM fatal o no fatal, reestenosis de angioplastia transluminal percutánea y by-pass arteriocoronarios.
Se han llevado a cabo varios metaanálisis47–50 que incluyen tanto estudios prospectivos como retrospectivos, para demostrar la asociación de resistencia con el AAS y mayor riesgo de eventos vasculares. Todos confirmaron el incremento de riesgo de aparición de nuevos eventos cardiovasculares en los pacientes no respondedores al AAS. Se observa también que esta asociación es mayor en los estudios no prospectivos que en los prospectivos (OR: 3,12; IC del 95%: 2,40–4,06 frente a OR: 1,75; IC del 95%: 1,35–2,28; p=0,005)50.
Mecanismos relacionados con el fenómeno de resistencia al ácido acetilsalicílicoExisten determinados factores que pueden modificar la respuesta al AAS. Se pueden clasificar en factores que provocan que se alcancen niveles insuficientes del AAS en sangre (mecanismos farmacocinéticos) y factores que inducen una modificación en las vías metabólicas o dianas farmacológicas a través de las cuales el AAS ejerce su efecto antiagregante (mecanismos farmacodinámicos).
El tipo de formulación farmacéutica puede influir en la distinta biodisponibilidad del AAS. Alberts et al9 relacionan la “formulación galénica del AAS” con una menor respuesta al AAS. En los pacientes a los que se les administró la formulación entérica la resistencia hallada fue de un 65 frente un 25% en los que se les administró la formulación normal.
Las “interacciones” medicamentosas también son otro factor importante en la reducción de la efectividad del tratamiento. Se ha hallado un porcentaje mayor de resistencia entre pacientes que toman estatinas18. Feher et al51 observaron que los pacientes en los que se halló resistencia al AAS estaban en tratamiento con esta clase de fármacos hipolipemiantes en mayor porcentaje que los pacientes respondedores al AAS (el 52 frente al 38%; p<0,05). Establecieron, por tanto, que la toma de estatinas de forma concomitante con AAS se considera un factor de riesgo independiente para el desarrollo de resistencia al AAS (OR: 5,9; IC del 95%: 1,83–16,9; p<0,005).
Otro grupos de fármacos como los AINE, compiten con el AAS y bloquean el acceso del AAS a su lugar de unión en la enzima COX-1, disminuyendo así su acción antiagregante. Esta interacción se ha demostrado con el ibuprofeno y no, en cambio, con el diclofenaco y ciertos inhibidores selectivos de la enzima COX-2, como el rofecoxib52. También se ha demostrado que la ingesta de inhibidores de la bomba de protones reduce la biodisponibilidad del AAS por una mayor acción de las esterasas de la mucosa gastrointestinal sobre el AAS, lo que produce una menor absorción de éste53. Sin embargo, en otro estudio54 realizado posteriormente se observó que el omeprazol no afectaba de forma significativa a la biodisponibilidad del AAS, así como al efecto antiagregante plaquetario. Por tanto, parece ser que se trata de una interacción de escasa relevancia clínica a pesar del uso conjunto muy frecuente de ambos fármacos en la práctica clínica.
Se han descrito “rutas alternativas de formación de TXA2” a través de la enzima COX-2, además de la vía plaquetaria en la que interviene la enzima COX-156,57. La enzima COX-2 es sintetizada principalmente en células nucleadas, como los monocitos, los macrófagos y nuevas plaquetas formadas en respuesta a estímulos inflamatorios, las cuales son capaces de sintetizar rápidamente esta enzima44. Se requieren dosis mayores del AAS y mayor frecuencia de administración para inhibir esta isoenzima y así producir un efecto antiinflamatorio, analgésico y antipirético. Se cree que en determinadas situaciones inflamatorias las células nucleadas podrían sintetizar determinadas cantidades de PGH2, precursora del TXA2, a través de la enzima COX-2, la cual no es inhibida en las dosis de AAS empleadas habitualmente para conseguir un efecto antiagregante44,55. Esta misma síntesis podría suceder a partir de la enzima COX-2 expresada en las plaquetas56.
Por otra parte, la PGH2 es precursora de PGD2, PGF2α y PGI23. La PGI2 presenta una acción vasodilatadora y antiagregante. Se ha postulado que dosis elevadas de AAS podrían inhibir suficientemente la producción de PGI2 como para atenuar su efecto antiagregante, aunque no ha podido demostrarse todavía de una forma convincente58.
Zimmermann et al59 también demuestran una inducción de la isoforma de la enzima COX-2 plaquetaria inmunorreactiva en pacientes que han sido sometidos a un by-pass arteriocoronario y son tratados con 100mg de AAS. En otro ensayo anterior se demuestra que un “aumento del turnover plaquetario”60 después del by-pass arteriocoronario puede explicar la inefectividad de bajas dosis de AAS, ya que la síntesis de TXA2 plaquetario debe ser bloqueada al menos en un 10% para una inhibición plaquetaria de forma eficiente. Por tanto, el AAS, de corta semivida plasmática, no sería capaz de inhibir esta síntesis en las nuevas plaquetas formadas.
De la misma forma, se postula la existencia de mutaciones genéticas que pueden explicar el fenómeno de resistencia al AAS. Se han descrito “polimorfismos o mutaciones en el gen de la enzima COX-1” que puedan favorecer la resistencia al AAS61.
Asimismo, se han descrito diferencias en la actividad del “receptor de fibrinógeno GPIIb/IIIa en relación con el polimorfismo del alelo P1A1,A2”62 que codifica la subunidad GPIIIa de dicho receptor. De esta forma, aquellas plaquetas homocigotas para el alelo P1A1 se han relacionado con una mayor resistencia al AAS62,63 a diferencia de las plaquetas que presentan al menos un alelo P1A2. Sin embargo, otros autores discuten estas conclusiones, encontrando una mayor resistencia al efecto del AAS en los pacientes portadores de al menos un alelo P1A2, así como un mayor riesgo de eventos trombóticos64–67.
Otros estudios relacionan niveles elevados en sangre del “factor de Von Willebrand”68, de “P-selectina”30 o de “ADP”69 con mayor resistencia al AAS. Una mayor sensibilidad de las plaquetas al “colágeno”70 también puede estar relacionada con la resistencia al AAS.
Por otra parte, se han encontrado niveles elevados del isoprostano “8-iso-PGF2α” en pacientes con angina inestable71 y fumadores72,73. Estos compuestos, similares a la PGF2, se originan a partir del AA a través de la peroxidación lipídica catalizada por radicales libres de oxígeno. Se trata de potentes vasoconstrictores que amplifican la respuesta de las plaquetas y, por tanto, podrían estar implicados en la patogénesis de las enfermedades cardiovasculares. De esta forma, muchos estudios demuestran una asociación significativa18,20,40,74-76, o una tendencia, pero no significativa37 entre la resistencia al AAS y el “hábito de fumar”. Sin embargo, se han encontrado resultados contradictorios. Davis et al74 llevan a cabo un ensayo aleatorizado doble ciego en 30 fumadores con enfermedad arteriocoronaria en el que se compara el efecto de 150mg, 300mg de AAS o placebo sobre el ratio de agregación plaquetaria antes y después de fumar. En el grupo de placebo el ratio de agregación plaquetaria hallado fue de 0,77 y de 0,72 antes y después de fumar, valores que no se alteraron tras la administración de ambas dosis de AAS.
Dentro de los posibles factores de riesgo no modificables, se ha relacionado el “sexo femenino” con la resistencia al AAS9,21,25,35,39. Otros autores han encontrado una posible relación con la “edad”9,22,25.
Se han descrito determinadas situaciones clínicas en las que existe un mayor nivel de reactividad plaquetaria. De esta forma, la “dislipemia y obesidad”18,21,37,38,40, así como la “diabetes” de tipo I y II37,38,40,77–80, se pueden acompañar de una menor respuesta al efecto antiagregante. En algunos de estos estudios esta tendencia no es estadísticamente significativa, quizás por el pequeño tamaño muestral, con lo que resulta difícil llegar a resultados concluyentes. Estos datos sugieren que puede existir asociación entre el síndrome metabólico y la resistencia al tratamiento antiplaquetario.
También se ha asociado la “hipertensión arterial”16,38 como factor de riesgo y se ha descrito casi el doble de resistencia al AAS en pacientes con síndromes coronarios agudos complicados con neumonía respecto a aquéllos sin “complicaciones respiratorias”81.
Asimismo, la liberación de catecolaminas en ciertas situaciones, como el “estrés” o el “ejercicio”19,82,83, produce una activación plaquetaria y disminuye la efectividad del AAS.
Se observa también una mayor resistencia al AAS en pacientes con “enfermedad cardíaca isquémica”18,40 e “insuficiencia cardíaca congestiva”38.
Manejo de pacientes con resistencia al ácido acetilsalicílicoEl manejo clínico de este fenómeno está aún en investigación. Aunque la medida de la agregación plaquetaria por test bioquímicos es una opción para detectar inicialmente a los pacientes no respondedores al AAS, no se ha estandarizado hoy en día cuál es el método más adecuado para identificar a estos pacientes, así como la correlación de los resultados obtenidos con la aparición de eventos cardiovasculares y su relación coste-efectividad. Por tanto, hoy en día no se recomienda su empleo como estrategia diagnóstica en la práctica clínica habitual.
Sin embargo, ante una sospecha de inefectividad al tratamiento con AAS, el primer paso es descartar una falta de “adherencia” antes que diagnosticar a un paciente como resistente, aun con resultados indicativos de ello según las pruebas de agregación plaquetaria17,31,84. Si, por el contrario, el paciente es un buen cumplidor, y se descartan otros motivos de inefectividad del tratamiento (como por ejemplo la presencia de interacciones medicamentosas), se podrían contemplar las siguientes opciones para contrarrestar este fenómeno, así como las consecuencias clínicas que se derivan de ello.
Aumento de dosis del ácido acetilsalicílicoExiste evidencia de que dosis de 75 a 150mg son tan eficaces en inhibir la agregación plaquetaria y disminuir eventos cardiovasculares2 como dosis mayores, con la ventaja de producir menos efectos adversos gastrointestinales ya que estos son dosis-dependiente.
Sin embargo, varios estudios sugieren que un aumento de dosis5,9,11,14,15,25,40,85 podría ser suficiente para el manejo clínico de aquellos pacientes resistentes al tratamiento o con presencia de factores de riesgo que pueden hacer sospechar una posible resistencia.
Un aspecto importante para tener en cuenta es que un aumento de dosis conlleva una mayor tasa de efectos adversos, en especial gastrointestinales y, por consecuencia, de posibles abandonos del régimen terapéutico. Se recomienda, por tanto, prescribir siempre la dosis mínima que sea efectiva2.
Adición de otros fármacos antiagregantesLa adición de una tienopiridina, como clopidogrel, ticlopidina (actualmente en desuso por producir mayor incidencia de neutropenia y supresión de médula ósea) o los nuevos antagonistas orales del receptor GPIIb/IIIa, actúa bloqueando otras rutas alternativas de agregación plaquetaria diferentes al AAS.
Las tienopiridinas inhiben la unión del ADP a su receptor plaquetario y, por tanto, la activación del complejo GPIIb/IIIa mediada por ADP, quedando inhibida de esta forma la agregación plaquetaria por otra vía distinta a la empleada por el AAS. La adición de clopidogrel al AAS ha demostrado ser superior al tratamiento en monoterapia con AAS en las indicaciones de síndrome coronario agudo sin elevación del segmento ST86, incluyendo pacientes sometidos a una intervención coronaria percutánea con colocación de stent87 y síndrome coronario agudo con elevación del segmento ST88. Por tanto, la adición de otro antiagregante plaquetario al tratamiento con AAS podría constituir una estrategia de tratamiento en pacientes resistentes al AAS.
Sin embargo, esta opción de doble antiagregación no ha sido específicamente evaluada en ningún ensayo clínico. Por otra parte, no hay que olvidar que también se ha observado el desarrollo de resistencia al clopidogrel, asociada con un mayor riesgo de eventos aterotrombóticos89–91 y que además puede ser a ambos fármacos (clopidogrel y AAS)92.
DiscusiónSe ha encontrado una gran variabilidad en la prevalencia de resistencia al AAS en los estudios revisados. Esta dispersión se puede explicar, entre otros motivos, por la falta de uniformidad encontrada en el método para definir la resistencia al AAS, la cual se mide a través de diferentes pruebas de agregación plaquetaria. Asimismo, aun empleando la misma técnica, no existe uniformidad en el valor de referencia empleado a partir del cual clasifican a los pacientes como resistentes o no respondedores al AAS.
Los métodos que se emplean para medir la agregación in vitro son mayoritariamente los siguientes: agregometría óptica, PFA-100 y RPFA. Harrison et al8 observaron una mayor sensibilidad para detectar pacientes resistentes con los métodos PFA-100 y RPFA. Sin embargo, resaltó la falta de correlación hallada entre estas pruebas frente a agregometría óptica (κ=0,16; IC del 95%: −0,08 a 0,39; p=0,11 y κ=0,09; IC del 95%: −0,12 a 0,30; p=0,32, respectivamente), así como entre ambas (κ=0,14; IC del 95%: −0,08 a 0,36; p=0,15). Otros estudios confirman también la escasa concordancia entre los resultados de distintos métodos y la capacidad diagnóstica del fenómeno denominado resistencia al AAS93,94.
Hay que considerar también que el diagnóstico de un paciente como resistente al AAS según una técnica que mide los efectos de ésta sobre el AA no es del todo preciso, puesto que ha postulado la existencia de otros mecanismos cardioprotectores del AAS independientes de esta vía de activación plaquetaria5.
Pero a pesar de que se hayan encontrado muchas dificultades para definir que un paciente es resistente al AAS desde un punto de vista bioquímico, según la mayoría los estudios, esta falta de respuesta se traduce en la disminución de su eficacia protectora frente a eventos cerebrovasculares y cardiovasculares.
Sin embargo, aunque parece lógica esta asociación, no está del todo establecida la relevancia clínica de este fenómeno. Hay que tener en cuenta también las siguientes limitaciones de los ensayos clínicos: pequeño tamaño muestral, diferente tiempo transcurrido desde la ingesta del AAS y toma de muestra para realizar el estudio de resistencia y heterogeneidad de la población incluida quienes pueden requerir un grado distinto de agregación plaquetaria para conseguir el efecto protector del AAS. En este contexto, es conocido el estado protrombótico que se puede dar en diferentes situaciones clínicas, como síndrome coronario agudo, en el que es necesaria una mayor antiagregación para contrarrestar esta situación clínica.
Otro factor para tener en cuenta es el cumplimiento terapéutico en los ensayos clínicos. En los estudios en los que no se haya controlado esta variable13,23,27,40,43–45 la asociación entre una actividad antiagregante disminuida con el fenómeno de resistencia al AAS y aparición de eventos clínicos puede quedar sobredimensionada en muchos de los casos.
Adicionalmente, nos cuestionamos si la resistencia al AAS es variable o es un valor absoluto en el tiempo.
Por último, hay que considerar que la trombosis inducida por las plaquetas no es el único mecanismo implicado en los procesos aterotrombóticos. Se ha demostrado que el tratamiento crónico con AAS se asocia con una reducción progresiva de la sensibilidad plaquetaria a ésta y, por tanto, una menor eficacia del tratamiento95.
La identificación de pacientes resistentes al tratamiento antiagregante sería recomendable en la práctica clínica. Sin embargo, al no existir un método de diagnóstico de referencia, todavía no se puede recomendar el empleo de rutina de estas técnicas.
Asimismo, la identificación de los factores que pueden influir en una disminución de la efectividad del tratamiento es importante para ayudar a optimizarlo de una forma individual. Las estrategias por seguir para contrarrestar esta antiagregación disminuida pueden ser las siguientes: aumento de dosis del AAS, terapia dual junto con otros agentes antiplaquetarios o modificación de determinados factores de riesgo, tales como la obesidad, la diabetes y el hábito de fumar. Sin embargo, estas medidas no han sido ampliamente investigadas en esta población de pacientes resistentes.
Es necesario el desarrollo de más estudios que aclaren todos los supuestos anteriores para establecer recomendaciones más claras para el manejo clínico de la resistencia al AAS de forma que se pueda conseguir el objetivo terapéutico del tratamiento con este antiagregante plaquetario en todos los pacientes de riesgo cardiovascular elevado.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

