

Desarrollar y validar internamente un modelo farmacocinético poblacional para doxorubicina y evaluar su capacidad predictiva en la personalización de su dosificación en pacientes oncológicos.
MétodosLas concentraciones plasmáticas de doxorubicina se determinaron en treinta y tres pacientes oncológicos, y se utilizó un modelo farmacocinético tricompartimental implementado en el programa NONMEN VI para determinar los parámetros farmacocinéticos. La identificabilidad de los parámetros se evaluó mediante bootstrap paramétrico, y la validación interna del modelo se realizó mediante bootstrap no-paramétrico, visual predictive check y numerical predictive check. La capacidad predictiva del modelo final se evaluó en términos de exactitud y precision de las concentraciones plasmáticas durante el primer y segundo ciclo de quimioterapia.
ResultadosEl aclaramiento de doxorubicina es de 58,8 l/h, con una variabilidad interpaciente del 29,2% y una variabilidad intrapaciente del 18,9%. El volumen de distribución estimado en estado estacionario es de 2.294 l, con unas variabilidades inter e intrapaciente del 7,3% y 26,1%, respectivamente. La validación interna confirma que el modelo farmacocinético poblacional resulta adecuado para describir la evolución temporal de las concentraciones plasmáticas de doxorubicina y su variabilidad en la población de estudio. La exactitud y la precisión de la predicción a posteriori de las concentraciones plasmáticas de doxorubicina mejoran un 63% y un 41% respecto a la predicción a priori.
ConclusiónEl modelo bayesiano poblacional desarrollado caracteriza adecuadamente la evolución temporal de las concentraciones plasmáticas de doxorubicina en la población de estudio y puede utilizarse de forma exacta y precisa para optimizar las pautas posológicas de doxorubicina en pacientes oncológicos.
To develop and internally validate a population pharmacokinetic model for doxorubicin and to evaluate its predictive performance for dose individualization in cancer patients.
MethodsDoxorubicin plasma concentrations were determined in thirty-three cancer patients treated with intravenous doxorubicin. A three-compartment pharmacokinetic model was implemented in the NONMEN VI programme to determine the doxorubicin pharmacokinetic parameters. The identifiability of the parameters was assessed by parametric bootstrap and model validation was performed using nonparametric bootstrap, visual predictive check, and numerical predictive check. The final model‘s predictive performance was evaluated in terms of accuracy and precision of plasma concentration predictions during the first and second cycles of chemotherapy.
ResultsDoxorubicin clearance was 58.8L/h, with interpatient variability of 29.2% and intrapatient variability of 18.9%. The estimated volume of distribution at steady state was 2294L, with inter-and intrapatient variability of 7.3% and 26.1%, respectively. Internal validation confirmed that the population pharmacokinetic model is appropriate to describe the time course of the doxorubicin plasma concentrations and its variability in this population. The accuracy and precision of an a posteriori prediction of doxorubicin plasma concentrations improved by 63% and 41% compared to the a priori prediction.
ConclusionThe Bayesian population pharmacokinetic model characterised the time course of doxorubicine plasma concentrations and can be accurately and precisely used to optimise doxorubicine dosing regimens in cancer patients.
Doxorubicina es un fármaco antineoplásico de amplio espectro que ejerce su efecto citotóxico al actuar en la replicación, la transcripción y la fragmentación del ADN. Además, doxorubicina también ejerce un efecto citotóxico mediante estrés oxidativo que conduce a apoptosis celular1,2. Está indicada en diferentes tumores sólidos (carcinoma de mama, carcinoma microcítico de pulmón, carcinoma gástrico y tumores ginecológicos, entre otros), tumores hematológicos (linfomas, leucemias y mieloma múltiple), sarcomas y tumores sólidos pediátricos3. Entre las pautas posológicas de doxorubicina más empleadas en monoterapia está la administración intravenosa de 60-75mg/m2 cada 3 semanas en perfusión de 30-60min de duración. Las pautas posológicas empleadas en combinación con otros agentes varían según protocolo entre 40-60mg/m2 cada 3 o 4 semanas4.
En las dosis habitualmente empleadas en clínica, doxorubicina presenta una farmacocinética lineal5 con una amplia distribución a tejidos periféricos que le confiere un volumen de distribución en estado estacionario alrededor de 800 l/m2, a pesar de su elevada unión a proteínas plasmáticas (79-85%), principalmente a la albúmina3,5. El volumen de distribución determinado en compartimento central oscila alrededor de 20 l, con una variabilidad interindividual cercana al 20%6,7. Doxorubicina no atraviesa la barrera hematoencefálica, pero sí la placenta, y es metabolizada ampliamente en el hígado por la enzima aldoketo reductasa que origina su metabolito principal, el doxorubicinol. También, aunque en menor medida, doxorubicina es sustrato de la isoforma 3A4 del citocromo P450 y el metabolito que origina es la doxorubicina-7-desoxiaglicona. Doxorubicinol tiene aproximadamente el 10% de la actividad de doxorubicina6, y junto con el metabolito 7-desoxiaglicona son los principales responsables de la toxicidad de doxorubicina8,9. Se estima que el aclaramiento sistémico de doxorubicina está alrededor de 55 l/h, con una variabilidad interindividual cercana al 20%6. El aclaramiento hepatobiliar, alrededor de 25 l/h, supone aproximadamente el 50% del aclaramiento de doxorubicina y coincide con la fracción de dosis eliminada por bilis en los 7 días sucesivos al tratamiento. Por el contrario, su aclaramiento renal es bajo, alrededor de 7 l/h, y tan solo un 12% de la dosis se recupera en orina después de 6 días desde el inicio del tratamiento5. El aclaramiento es menor en pacientes de edad avanzada10,11, en obesos12 y en mujeres13. El tipo de tumor también influye en el aclaramiento de doxorubicina por cuanto que pacientes con cáncer de mama han presentado un aclaramiento de doxorubicina de 26 l/h, inferior al presentado por mujeres con tumores de diferente etiología cuyo valor es de 53 l/h13. Además, existe una correlación entre valores elevados de biomarcadores de funcionalidad hepáticos y valores reducidos de aclaramiento de doxorubicina. Estos datos han sugerido la reducción de un 50 y 75% de la dosis de doxorubicina en pacientes con unos valores elevados de bilirrubina o aspartato aminotransferasa (AST), respectivamente14.
Entre los efectos adversos más importantes que limitan las dosis de doxorubicina utilizadas en la práctica clínica se encuentran la toxicidad cardíaca y la hematológica, aunque también se ha documentado toxicidade gastrointestinal, dermatológica, hepática y renal. El efecto cardiotóxico es consecuencia de la síntesis y acumulación de sus metabolitos en los cardiomiocitos. Estudios in vitro determinaron que doxorrubicinol inhibe la contractilidad del músculo cardiaco de ratón con una potencia entre 10 y 30 veces mayor que doxorubicina8,15. A concentraciones de 50μg/ml de doxorubicinol y de 350μg/ml de doxorubicina se produce una reducción de la contractilidad del 69 y 11%, respectivamente16,17. El desarrollo de cardiotoxicidad crónica está relacionado con la concentración plasmática máxima (Cmáx) de doxorubicina. Así, la cardiomiopatía puede ser reducida significativamente prolongando la duración de la infusión4,7. Este tipo de toxicidad también está relacionado con dosis altas acumuladas, dosis superiores a 550 y 600mg/m2 se relacionan con incidencias de cardiotoxicidad del 18 y 36%, respectivamente18, y por tanto, la dosis total acumulada de doxorubicina no debe exceder los 550mg/m2.
Recientemente se ha determinado la relación entre la exposición de doxorubicina y su eficacia clínica en pacientes afectados de linfoma de Hodgkin. De los 19 pacientes evaluados, 14 presentaron respuesta completa durante al menos 12 meses de tratamiento con doxorubicina y se asociaron con valores más elevados de área bajo la curva (AUC) y Cmáx respecto a los pacientes que no obtuvieron respuesta al tratamiento (36,3 vs 19,3 ngmin/ml, p=0,08; 280 vs 73ng/ml, p=0,06)19.
La monitorización de las concentraciones plasmáticas de doxorubicina se justifica principalmente por 3 razones; a) es un fármaco con un margen terapéutico estrecho; b) presenta una elevada variabilidad en su farmacocinética6,20, y c) existe correlación entre sus concentraciones en plasma y eficacia19 y toxicidad4,7,8,15. Las características farmacocinéticas y farmacodinámicas de doxorubicina justifican la monitorización de sus concentraciones plasmáticas aunque su implementación rutinaria en la práctica clínica es escasa debido a la necesidad de disponer de una técnica analítica que presente adecuada precisión, exactitud, especificidad y selectividad para la determinación de doxorubicina en muestras biológicas de pacientes21.
La metodología bayesiana es una de las técnicas más empleadas para la individualización posológica22. La aplicación de esta técnica con garantía de éxito requiere la estimación exacta y precisa de los parámetros farmacocinéticos a partir de los datos correspondientes a la población específica de pacientes subsidiarios de ajuste posológico23. La farmacocinética de doxorubicina se ha descrito en la literatura médica mediante modelos farmacocinéticos lineales basados en dos20,24 y tres6,7 compartimentos, y se ha utilizado el modelo de efectos mixtos para caracterizar tanto la tendencia central como la variabilidad inter e intraindividual en los parámetros cinéticos de doxorubicina, así como sus relaciones con covariables demográficas y clínicas6,25. Por ello, el objetivo del presente trabajo ha sido desarrollar y validar internamente un modelo farmacocinético poblacional para doxorubicina y evaluar su capacidad predictiva en la personalización de su dosificación en pacientes oncológicos.
MétodosCriterios de selección de pacientesSe incluyeron en el estudio pacientes oncológicos adultos subsidiarios de recibir tratamiento con doxorubicina, con edad comprendida entre 18 y 80 años, con una expectativa de vida superior a 3 meses, función medular normal (recuento de leucocitos>4×109/l, hemoglobina > 10 g/dl, recuento de neutrófilos>1,5×109/l y recuento de plaquetas>150×109/l), función renal normal (creatinina sérica<1,5mg/dl), función cardiaca normal (confirmada por electrocardiograma y fracción de eyección ventricular [FEV]>50%) y una función hepática conservada (bilirrubina<1,2mg/dl, AST y alanina aminotransferasa (ALT) ≤ 40 U/l). Se excluyeron del estudio las mujeres lactantes o en periodo gestacional, pacientes con estado general mayor de 2 según la escala ECOG, o inferior a 60% en la escala Karnofsky, pacientes que hubiesen recibido radioterapia extensa previa, pacientes con trastornos psquiátricos o de cualquier otro tipo que afectase a la capacidad para otorgar el consentimiento informado verdadero para la participación en el estudio, pacientes que hubiesen presentado cualquier circunstancia que impidiese que el tratamiento y su seguimiento se realizase según protocolo establecido, como puede ser la progresión de enfermedad o la aparición de cardiotoxicidad y aquellos pacientes que hubiesen participado en un estudio clínico al menos 30 días antes.
El estudio se llevó a cabo según los principios de experimentación humana establecidos en la Declaración de Helsinki y fue aprobado por el Comité Ético Investigador del Hospital. En todos los casos se obtuvo el consentimiento informado del paciente tras una entrevista personal donde se le informó verbalmente y por escrito de los potenciales riesgos y beneficios del tratamiento y el estudio.
Toma de muestras y técnica analíticaEn los primeros 2 ciclos de quimioterapia se tomaron muestras sanguíneas a las 0,75, 1, 2 y 5,5 h desde el inicio de la perfusión. Estos tiempos fueron seleccionados según la teoría de muestreo óptimo y los modelos farmacocinéticos previamente publicados en la literatura biomédica. Las muestras de sangre se extrajeron del brazo contralateral al que se administró la quimioterapia y se recogieron en tubos de plástico con heparina de litio como anticoagulante (Sarstedt®). Las muestras se centrifugaron durante 10min a 3.500rpm y el plasma se congeló a −80°C hasta su valoración.
La determinación de concentraciones plasmáticas de doxorubicina se realizó mediante cromatografía líquida de alta resolución acoplada a detección de fluorescencia según una técnica analítica publicada con anterioridad21. Previamente al análisis, las muestras plasmáticas se sometieron a una extracción sólido-líquido con la que se obtuvo un rendimiento superior al 79%. Como fase estacionaria se empleó una columna cromatográfica Kromasil C18 y como fase móvil se empleó una mezcla de dihidrogenofosfato potásico 0,05M (pH 3,55) y acetonitrilo en proporción 65:35 (v/v). La elusión de los analitos fue isocrática y se realizó a un flujo de 0,6ml/min. Las longitudes de onda de emisión y excitación seleccionadas fueron de 480nm y 560nm, respectivamente. La técnica analítica empleada permitió determinar concentraciones plasmáticas de doxorubicina entre 1 y 900ng/ml con adecuada exactitud (error relativo < 4%) y precisión (CV < 7%). La exactitud y precision para el límite de cuantificación de 1ng/ml fueron inferiores al 1 y 5%, respectivamente.
Análisis farmacocinéticoProgramas informáticosEl análisis farmacocinético se realizó en el programa informático NONMEM VI versión 2.0 (ICON, Hanover, MD, EE.UU.)26 con la aproximación de primer orden condicional (FOCE) con interacción. La compilación del programa se realizó con el programa DIGITAL Visual Fortran versión 6.6C. Los gráficos y el resto de análisis estadísticos, incluida la evaluación de los modelos ajustados con NONMEM, se realizaron con el programa S-Plus 6.1 Professional Edition para Windows (Insightful, Seattle, WA, EE.UU.).
Modelo farmacoestadísticoTras el análisis exploratorio gráfico de los datos obtenidos y la evaluación de la literatura científica, se seleccionó un modelo farmacocinético tricompartimental, con distribución y eliminación lineal. Este modelo se parametrizó en términos de aclaramiento (CL, L/h), volumen de distribución central (V1, L), flujo entre el compartimento central y el periférico liviano (Q2, L/h), volumen del compartimento periférico liviano (V2, L), flujo entre el compartimento central y el periférico profundo (Q3, L/h) y volumen del compartimento periférico profundo (V3, L).
Se asumió que la variabilidad inter e intraindividual seguían una distribución log-normal según la ecuación:
donde Pj,k es el parámetro farmacocinético individual para el paciente j-ésimo en la ocasión k-ésima, P* es el valor típico poblacional, ηj y τk son variables independientes y aleatorias que cuantifican las diferencias entre el valor típico poblacional y su valor individual en cada ocasión que se administra el tratamiento, y se asume que siguen una distribución normal de media cero y varianza ωP2 y πP2, respectivamente Las magnitudes de las variabilidades inter e intraindividuales determinadas en Cl, Q2 y V2 se expresaron aproximadamente los coeficientes de variación (CV). En el resto de parámetros farmacocinéticos, y de acuerdo con las publicaciones farmacocinéticas previas, no se evaluó la variabilidad interindividual ni la intraindividual. La variabilidad residual se evaluó con un modelo de error aditivo tras la transformación logarítmica de las concentraciones observadas y las correspondientes predicciones del modelo, de acuerdo con la ecuación:donde Cobs es la concentración observada de doxorubicina en plasma, Cpred es la concentración predicha por el modelo, y ¿ es una variable aleatoria independiente con distribución normal con media cero y varianza σ2, que describe la diferencia del logaritmo de la concentración observada y el logaritmo de la predicción del modelo, es decir, el valor residual.Análisis de datos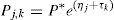
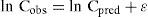
El análisis famacocinético de las concentraciones plasmáticas de doxorubicina se realizó mediante 3 métodos distintos. El método A se utilizó para estimar los parámetros farmacocinéticos poblacionales del modelo descrito a partir de las concentraciones de doxorubicina obtenidas en la muestra de pacientes estudiada. Este método, utilizado en la literatura científica para caracterizar los parámetros de los modelos farmacocinéticos de doxorubicina6, necesita un número de muestras por paciente elevado, circunstancia que habitualmente no es factible en la práctica clínica rutinaria, como sucedió en este caso. Por lo tanto, el muestreo reducido empleado en el presente estudio podría resultar insuficiente para estimar con exactitud y precisión todos los parámetros del modelo farmacocinético poblacional de 3 compartimentos. Ante esta situación, una alternativa (método B) fue fijar los parámetros Q2, V2, Q3 y V3 a los valores publicados por Callies et al.6. La otra alternativa utilizada fue el análisis bayesiano poblacional (método C). El método C se utilizó para actualizar los parámetros farmacocinéticos poblacionales del modelo de Callies et al.6 a partir de las concentraciones de doxorubicina obtenidas en la muestra de pacientes estudiada. En este caso, los parámetros de efecto fijo y aleatorio establecidos por Callies et al.6 se utilizaron como información a priori para la estimación bayesiana poblacional implementada en NONMEM mediante la subrutina $PRIOR con la opción NWPRI. Esta subrutina implementa una función de penalización derivada de los datos a priori mediante una distribución normal para los parámetros de efecto fijo y una distribución de Wishart invertida para los efectos aleatorios. Además, se definió la incertidumbre a priori de los parámetros de efecto fijo y los grados de libertad de los efectos aleatorios tal y como describen Gisleskog et al.27. Este método no incluye la información a priori del error residual.
Con objeto de evaluar el efecto que la estrategia de toma de muestras reducida tiene sobre la identificabilidad de los parámetros del modelo farmacocinético poblacional de doxorubicina se realizó un bootstrap paramétrico (PBT) de los métodos A, B y C. Así, se utilizaron los parámetros del modelo de Callies et al.6 para generar 100 bases de datos diferentes aunque con idéntica estructura a la base de datos original. Cada una de las réplicas de la base de datos original se analizó con los 3 métodos de análisis (A, B y C) y se obtuvieron los correspondientes parámetros farmacocinéticos poblacionales de tendencia central y variabilidad. A partir de estas estimaciones, se calculó la media, el error estándar y el intervalo de confianza del 95% de cada parámetro y se comparó con el valor del parámetro del modelo de Callies et al.6 utilizado en la simulación de las bases de datos. Si el parámetro farmacocinético es identificable a partir de la base de datos disponible y el método de análisis utilizado, el intervalo de confianza del 95% de cada parámetro debería incluir el valor del parámetro obtenido por Callies et al.6.
Validación del modeloLa validación interna del modelo se realizó mediante 3 técnicas diferentes y complementarias como son el bootstrap no paramétrico (NPBT), el visual predictive check (VPC) y el numerical predictive check (NPC). En el NPBT se consideró que el modelo no presentaba sesgos si las estimaciones de los parámetros del modelo desarrollado con la base de datos original se encontraban dentro del intervalo de confianza del 95% construido con bases de datos generadas mediante muestreo de pacientes con reemplazamiento con el programa WINGS para NONMEM VI (N. Holford, Version 616, Auckland, Nueva Zelanda). En el VPC se comparó la evolución temporal de concentraciones plasmáticas de doxorubicina correspondientes a la base de datos original con la media y el intervalo de predicción del 90% de concentraciones plasmáticas de doxorubicina simuladas a partir de la base de datos original y de los valores de los parámetros de efecto fijo y aleatorio estimados por el modelo final. Por último, en el NPC se estratificaron las concentraciones plasmáticas en 4 grupos en función del tiempo de muestreo (0,75, 1, 2 y 5,5 h desde el inicio de la perfusión) en cada una de la bases de datos simuladas con el modelo farmacocinético seleccionado. En cada grupo se calculó el valor medio y el CV (%) de las concentraciones plasmáticas de doxorubicina, así como los respectivos intervalos de confianza del 95% a partir de los percentiles 5, 50 y 95%. Estos resultados se compararon con la media y el CV (%) de las concentraciones observadas para cada tiempo.
Evaluación de la capacidad predictivaLa capacidad predictiva del modelo se evaluó en términos de exactitud y precisión de las concentraciones plasmáticas de doxorubicina según la teoría de errores de predicción de Sheiner y Beal28. Como parámetro de exactitud se calculó la media del error relativo medio de predicción (ERM) existente entre las concentraciones plasmáticas observadas y las correspondientes predicciones del modelo. Como parámetro de precisión se calculó la raíz cuadrada del error cuadrático medio de predicción (RECM). Los valores de ERM y RECM del modelo se calcularon mediante las ecuaciones 3 y 4, respectivamente:
donde, Qi representa la estimación de la concentración plasmática obtenida en el i-ésimo paciente y Q* representa el valor de la concentración plasmática obtenido en ese mismo paciente. Además, se comprobó si el ERM difería significativamente (p<0,05) del valor de cero a través del cálculo de los intervalos de confianza del 95% de dicho valor.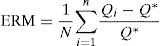
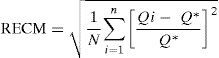
El análisis de la exactitud y la precisión se realizó por separado para los dos primeros ciclos de tratamiento. Las concentraciones plasmáticas predichas por el modelo para el primer ciclo de quimioterapia se obtuvieron según los parámetros típicos poblacionales (predicción a priori) puesto que no se disponía de información farmacocinética previa del paciente. Las concentraciones plasmáticas predichas por el modelo para el segundo ciclo de quimioterapia se generaron según los parámetros farmacocinéticos individuales del paciente obtenidos mediante estimación bayesiana individual a partir de las concentraciones plasmáticas observadas en el primer ciclo y el modelo poblacional desarrollado (predicción a posteriori). De esta forma es posible evaluar el efecto de la monitorización de las concentraciones plasmáticas de doxorubicina en la optimización farmacoterapéutica de doxorubicina en pacientes oncológicos.
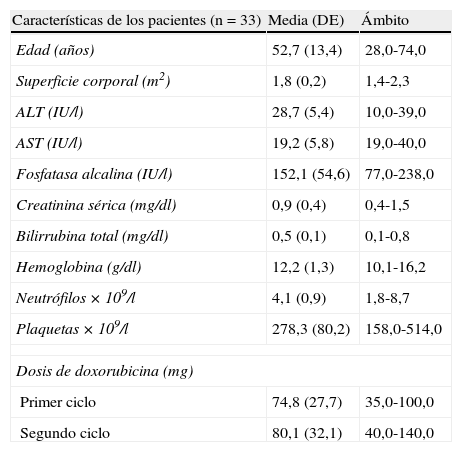
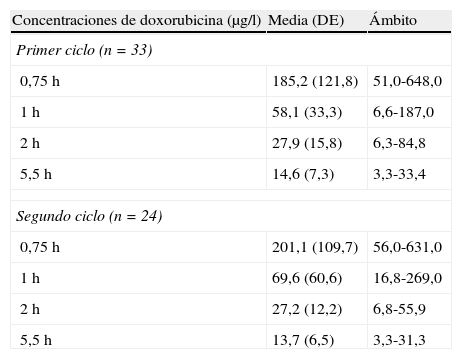
ResultadosEl presente estudio se ha realizado con un total de 33 pacientes oncológicos, de los cuales el 48,5% fueron varones y el 51,5% mujeres. Los pacientes recibieron tratamiento con doxorubicina intravenosa en un régimen de administración bisemanal, con una dosis media de 41,6mg/m2 (20-73mg/m2) y con una duración de la perfusión de entre 30 y 60min. La distribución de los pacientes según el tipo de tumor fue linfoma (n=10), carcinoma de mama (n=6), carcinoma de ovario (n=4), carcinoma de pulmón (n=4), carcinoma gástrico (n=3), sarcoma de partes blandas (n=2), vesical (n=2), renal (n=1) y carcinoma de cérvix (n=1). Doxorubicina se administró en terapia combinada con otros antineoplásicos entre los que se incluyeron ciclofosfamida (23%), irinotecán (16%), vincristina (13%), vinblastina (10%) o pemetrexed (5%), entre otros, según el tipo de tumor primario a tratar. Se monitorizaron un total de 57 ciclos de quimioterapia que supuso la cuantificación de la concentración plasmática de doxorubicina en 205 muestras. En la tabla 1 se resumen las características de la población al inicio del estudio. En la tabla 2 se resumen las concentraciones plasmáticas de doxorubicina en primer y segundo ciclo.
Resumen de las características basales de los pacientes y su tratamiento con doxorubicina
| Características de los pacientes (n=33) | Media (DE) | Ámbito |
| Edad (años) | 52,7 (13,4) | 28,0-74,0 |
| Superficie corporal (m2) | 1,8 (0,2) | 1,4-2,3 |
| ALT (IU/l) | 28,7 (5,4) | 10,0-39,0 |
| AST (IU/l) | 19,2 (5,8) | 19,0-40,0 |
| Fosfatasa alcalina (IU/l) | 152,1 (54,6) | 77,0-238,0 |
| Creatinina sérica (mg/dl) | 0,9 (0,4) | 0,4-1,5 |
| Bilirrubina total (mg/dl) | 0,5 (0,1) | 0,1-0,8 |
| Hemoglobina (g/dl) | 12,2 (1,3) | 10,1-16,2 |
| Neutrófilos × 109/l | 4,1 (0,9) | 1,8-8,7 |
| Plaquetas × 109/l | 278,3 (80,2) | 158,0-514,0 |
| Dosis de doxorubicina (mg) | ||
| Primer ciclo | 74,8 (27,7) | 35,0-100,0 |
| Segundo ciclo | 80,1 (32,1) | 40,0-140,0 |
ALT: alanino aminotransferasa; AST: aspartato aminotransferasa; DE: desviación estándar.
Concentraciones plasmáticas de doxorubicina en el primer y segundo ciclo
| Concentraciones de doxorubicina (μg/l) | Media (DE) | Ámbito |
| Primer ciclo (n=33) | ||
| 0,75h | 185,2 (121,8) | 51,0-648,0 |
| 1h | 58,1 (33,3) | 6,6-187,0 |
| 2h | 27,9 (15,8) | 6,3-84,8 |
| 5,5h | 14,6 (7,3) | 3,3-33,4 |
| Segundo ciclo (n=24) | ||
| 0,75h | 201,1 (109,7) | 56,0-631,0 |
| 1h | 69,6 (60,6) | 16,8-269,0 |
| 2h | 27,2 (12,2) | 6,8-55,9 |
| 5,5h | 13,7 (6,5) | 3,3-31,3 |
DE: desviación estándar.
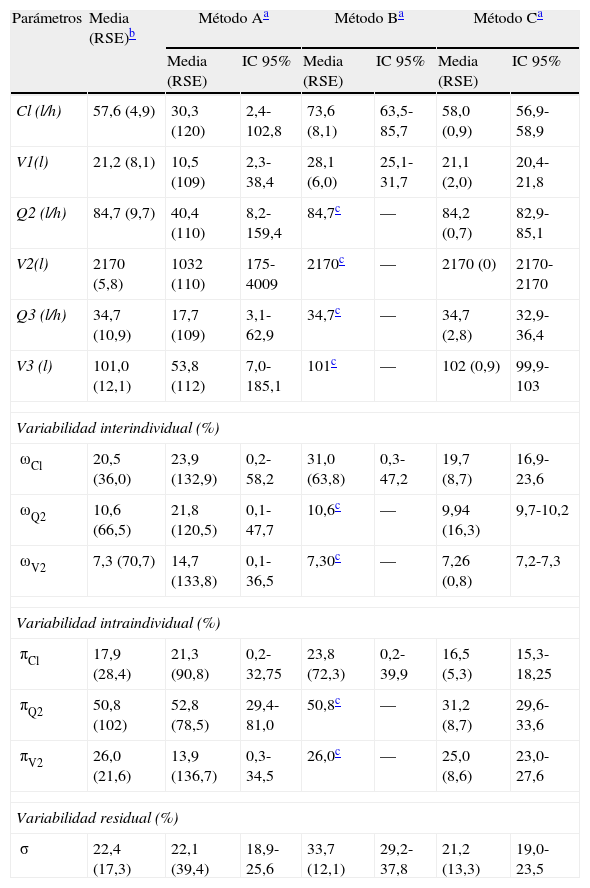
Los resultados del análisis farmacocinético no pueden interpretarse de manera aislada respecto a los resultados del PBT realizado con los métodos A, B y C para evaluar el efecto que la estrategia de toma de muestras reducida tiene sobre la identificabilidad de los parámetros del modelo farmacocinético poblacional de doxorubicina y que se recogen en la tabla 3. Con el método A la estimación de los parámetros farmacocinéticos de efecto fijo resultó ser un 50% menor que los valores referidos en la literatura médica6, y sus correspondientes errores estándar relativos (RSE) fueron superiores al 100%. Estos resultados evidencian la imposibilidad de identificar correctamente los parámetros del modelo tricompartimental a partir de los datos experimentales. No obstante, al fijar los parámetros farmacocinéticos de los compartimentos periféricos a los valores referidos en la literatura (método B), las estimaciones de Cl y V1 resultaron ser parecidas a los valores poblacionales referidos por Callies et al.6 y sus correspondientes RSE fueron inferiores al 8,1%. Sin embargo, los RSE de las variabilidades interindividuales de estos parámetros aumentaron hasta un 72,3%. Además, al fijar 4 parámetros de efecto fijo y otros 4 de efecto aleatorio se perdió capacidad predictiva por cuanto que el error residual aumentó hasta un 33,7%. Con el método C, tanto los parámetros de efecto fijo como los de efecto aleatorio son perfectamente identificables. Estos resultados permiten explicar las diferencias en los parámetros determinados con los 3 métodos.
Parámetros farmacocinéticos poblacionales y análisis bootstrap paramétricos realizados con los métodos A, B y C
| Parámetros | Media (RSE)b | Método Aa | Método Ba | Método Ca | |||
| Media (RSE) | IC 95% | Media (RSE) | IC 95% | Media (RSE) | IC 95% | ||
| Cl (l/h) | 57,6 (4,9) | 30,3 (120) | 2,4-102,8 | 73,6 (8,1) | 63,5-85,7 | 58,0 (0,9) | 56,9-58,9 |
| V1(l) | 21,2 (8,1) | 10,5 (109) | 2,3-38,4 | 28,1 (6,0) | 25,1-31,7 | 21,1 (2,0) | 20,4-21,8 |
| Q2 (l/h) | 84,7 (9,7) | 40,4 (110) | 8,2-159,4 | 84,7c | — | 84,2 (0,7) | 82,9-85,1 |
| V2(l) | 2170 (5,8) | 1032 (110) | 175-4009 | 2170c | — | 2170 (0) | 2170-2170 |
| Q3 (l/h) | 34,7 (10,9) | 17,7 (109) | 3,1-62,9 | 34,7c | — | 34,7 (2,8) | 32,9-36,4 |
| V3 (l) | 101,0 (12,1) | 53,8 (112) | 7,0-185,1 | 101c | — | 102 (0,9) | 99,9-103 |
| Variabilidad interindividual (%) | |||||||
| ωCl | 20,5 (36,0) | 23,9 (132,9) | 0,2-58,2 | 31,0 (63,8) | 0,3-47,2 | 19,7 (8,7) | 16,9-23,6 |
| ωQ2 | 10,6 (66,5) | 21,8 (120,5) | 0,1-47,7 | 10,6c | — | 9,94 (16,3) | 9,7-10,2 |
| ωV2 | 7,3 (70,7) | 14,7 (133,8) | 0,1-36,5 | 7,30c | — | 7,26 (0,8) | 7,2-7,3 |
| Variabilidad intraindividual (%) | |||||||
| πCl | 17,9 (28,4) | 21,3 (90,8) | 0,2-32,75 | 23,8 (72,3) | 0,2-39,9 | 16,5 (5,3) | 15,3-18,25 |
| πQ2 | 50,8 (102) | 52,8 (78,5) | 29,4-81,0 | 50,8c | — | 31,2 (8,7) | 29,6-33,6 |
| πV2 | 26,0 (21,6) | 13,9 (136,7) | 0,3-34,5 | 26,0c | — | 25,0 (8,6) | 23,0-27,6 |
| Variabilidad residual (%) | |||||||
| σ | 22,4 (17,3) | 22,1 (39,4) | 18,9-25,6 | 33,7 (12,1) | 29,2-37,8 | 21,2 (13,3) | 19,0-23,5 |
RSE: Error estándar relativo. IC: Intervalo de confianza.
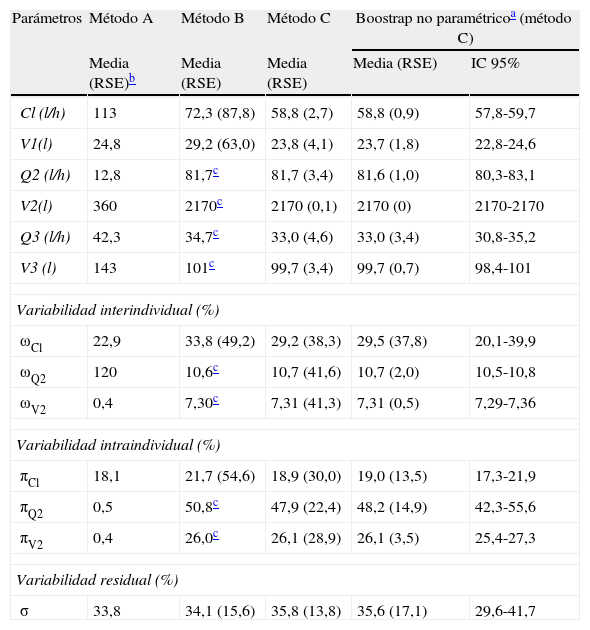
Los resultados del análisis farmacocinético de las concentraciones plasmáticas mediante el método de estimación bayesiano poblacional (métodoC) se muestran en la tabla 4 junto con los resultados del NPBT. Las estimaciones de los parámetros de efecto fijo y de efecto aleatorio son similares a las del modelo de Callies et al.6, y sus correspondientes RSE, son inferiores al 5 y al 42%, respectivamente. De las mil réplicas analizadas durante el NPBT, un 1,9% no minimizaron con éxito y se excluyeron del análisis. Las estimaciones poblacionales del modelo bayesiano fueron similares a la media de las réplicas generadas por el NPBT y estaban contenidas en el intervalo de confianza del 95%. Estos resultados demuestran la adecuada exactitud y precisión de los parámetros estimados mediante la metodología bayesiana poblacional. Únicamente cabe destacar que el valor de V2 no varía entre las réplicas bootstrap, circunstancia que claramente indica la falta de información en los datos obtenidos para poder estimar ese valor, y justifica la evaluación del método B y C como alternativa al método A.
Parámetros farmacocinéticos poblacionales y resultados del análisis bootstrap no paramétrico
| Parámetros | Método A | Método B | Método C | Boostrap no paramétricoa (métodoC) | |
| Media (RSE)b | Media (RSE) | Media (RSE) | Media (RSE) | IC 95% | |
| Cl (l/h) | 113 | 72,3 (87,8) | 58,8 (2,7) | 58,8 (0,9) | 57,8-59,7 |
| V1(l) | 24,8 | 29,2 (63,0) | 23,8 (4,1) | 23,7 (1,8) | 22,8-24,6 |
| Q2 (l/h) | 12,8 | 81,7c | 81,7 (3,4) | 81,6 (1,0) | 80,3-83,1 |
| V2(l) | 360 | 2170c | 2170 (0,1) | 2170 (0) | 2170-2170 |
| Q3 (l/h) | 42,3 | 34,7c | 33,0 (4,6) | 33,0 (3,4) | 30,8-35,2 |
| V3 (l) | 143 | 101c | 99,7 (3,4) | 99,7 (0,7) | 98,4-101 |
| Variabilidad interindividual (%) | |||||
| ωCl | 22,9 | 33,8 (49,2) | 29,2 (38,3) | 29,5 (37,8) | 20,1-39,9 |
| ωQ2 | 120 | 10,6c | 10,7 (41,6) | 10,7 (2,0) | 10,5-10,8 |
| ωV2 | 0,4 | 7,30c | 7,31 (41,3) | 7,31 (0,5) | 7,29-7,36 |
| Variabilidad intraindividual (%) | |||||
| πCl | 18,1 | 21,7 (54,6) | 18,9 (30,0) | 19,0 (13,5) | 17,3-21,9 |
| πQ2 | 0,5 | 50,8c | 47,9 (22,4) | 48,2 (14,9) | 42,3-55,6 |
| πV2 | 0,4 | 26,0c | 26,1 (28,9) | 26,1 (3,5) | 25,4-27,3 |
| Variabilidad residual (%) | |||||
| σ | 33,8 | 34,1 (15,6) | 35,8 (13,8) | 35,6 (17,1) | 29,6-41,7 |
RSE: Error estándar relativo. IC: Intervalo de confianza.
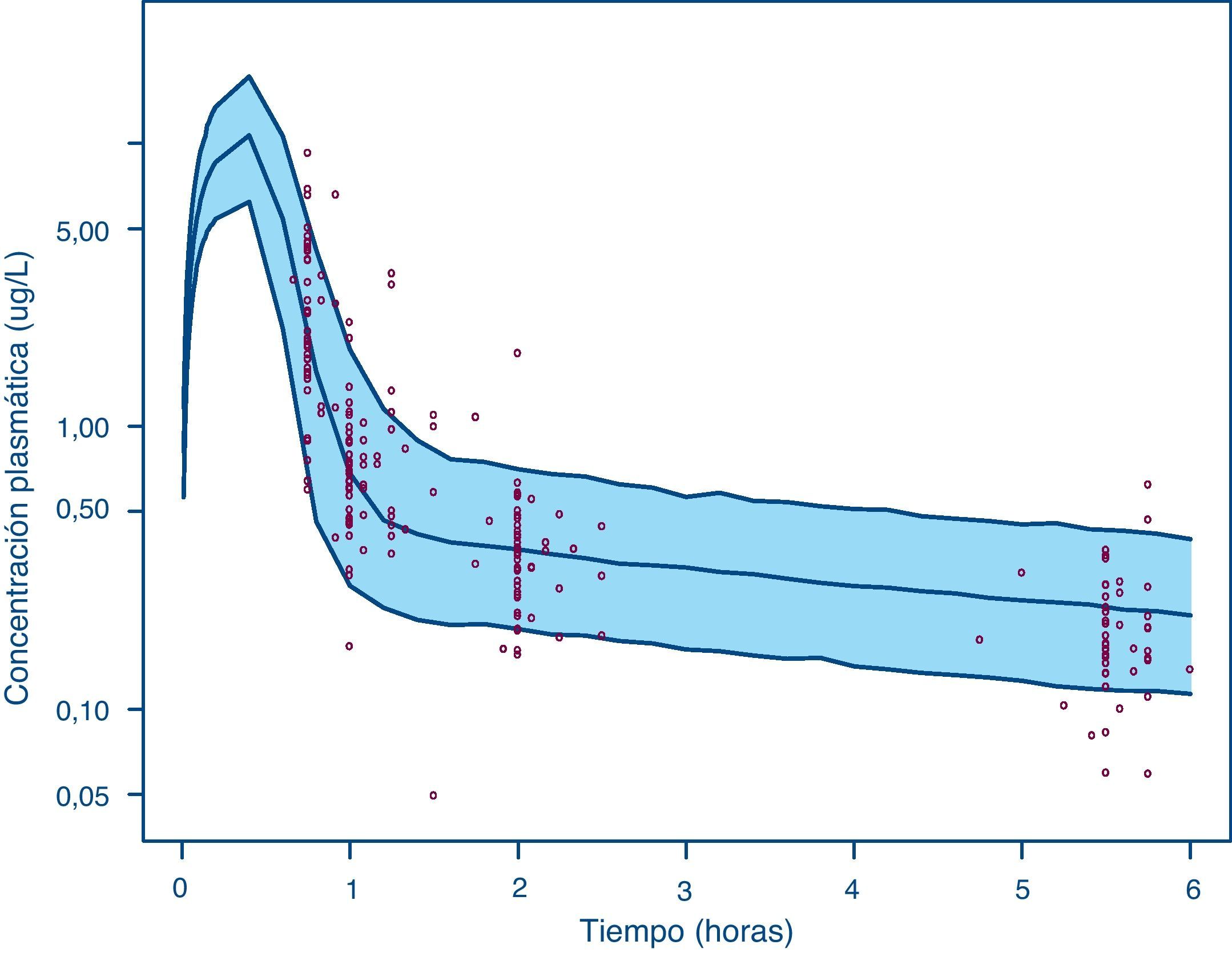
En la figura 1 se presentan los resultados del VPC, donde se evidencia que la evolución temporal de las concentraciones plasmáticas de doxorubicina y su variabilidad es caracterizada de forma adecuada. De hecho, un 12% del total de concentraciones experimentales del primer y segundo ciclo quedaron fuera del intervalo de predicción del 90% (p=0,342). Estos resultados confirman que el modelo describe adecuadamente los datos observados en la población de pacientes de estudio, tanto en el primer ciclo como en el segundo ciclo de tratamiento.

Visual predictive check de la evolución temporal de las concentraciones plasmáticas de doxorubicina en primer y segundo ciclo. Las líneas azules continuas representa los percentiles 5, 50 y 95, respectivamente, en la predicción de concentraciones, el área azul sombreada representa el intervalo de predicción del 90% y los puntos rojos representan las concentraciones de doxorubicina observadas en plasma durante los primeros 2 ciclos de quimioterapia.
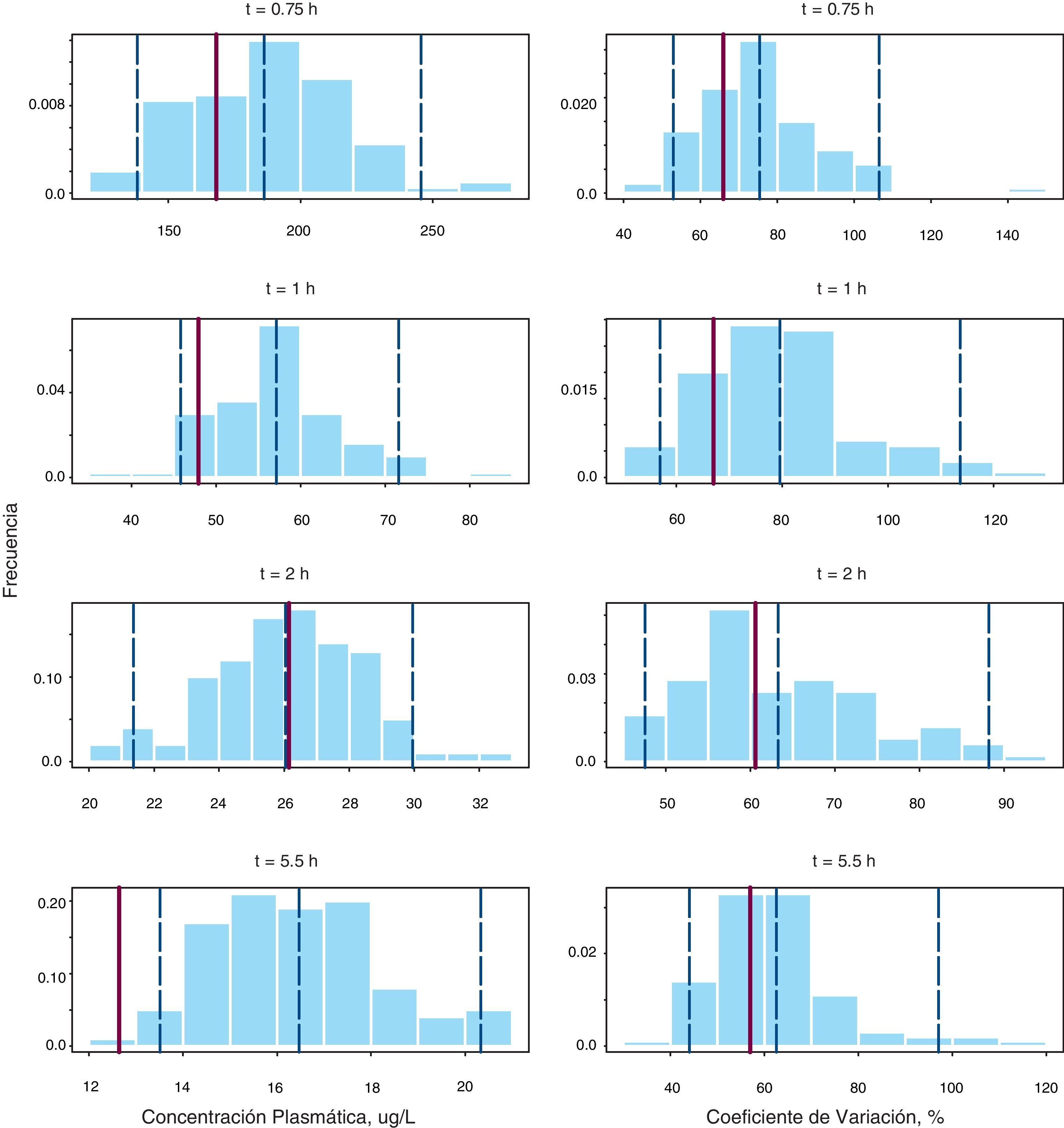
La figura 2 recoge los resultados del NPC para los 2 parámetros evaluados en cada tiempo de muestreo: las concentraciones plasmáticas y sus correspondientes CV (%). En todos los paneles las barras del histograma representan la densidad de probabilidad de los datos simulados, la línea vertical punteada azul representa los percentiles 5, 50, y 95 de la distribución simulada y la línea vertical roja representa el valor del parámetro obtenido a partir de la base de datos original. Como se puede observar, las concentraciones medias en la población estudiada se incluyen dentro del intervalo de confianza del 95% en todos los casos, excepto para el tiempo de 5,5 h donde el valor observado, 12,6μg/l, fue ligeramente inferior al límite inferior del IC 95% estimado, 13,5μg/l. En cuanto al CV (%) en todos los casos la media observada se encuentra dentro del intervalo de confianza. Por tanto, estos resultados en su conjunto confirman la validez del método C para describir tanto las concentraciones plasmáticas de doxorubicina como su variabilidad.
 junto con sus coeficientes de variación (paneles de la derecha). Las líneas verticales discontinuas representan los percentiles 5, 50 y 95 de la distribución de concentraciones simuladas y la línea vertical roja continua representa la correspondiente observada en la muestra de paciente analizada, respectivamente.")
Numerical predictive check para la media de concentraciones plasmáticas de doxorubicina a cada uno de los tiempos de recogida de muestra (paneles de la izquierda) junto con sus coeficientes de variación (paneles de la derecha). Las líneas verticales discontinuas representan los percentiles 5, 50 y 95 de la distribución de concentraciones simuladas y la línea vertical roja continua representa la correspondiente observada en la muestra de paciente analizada, respectivamente.
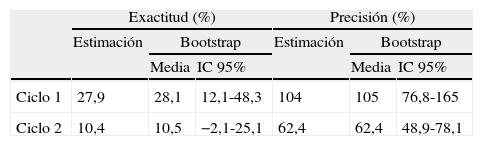
Por último, los resultados de la valoración de la capacidad predictiva del modelo seleccionado se resumen en la tabla 5. La predicción de las concentraciones plasmáticas de doxorubicina a priori presentó un sesgo sistemático ya que el IC 95% no incluyó el valor cero. La información individual del paciente, obtenida tras la monitorización de las concentraciones plasmáticas de doxorubicina en el primer ciclo de quimioterapia, añadió valor en la capacidad predictiva del modelo por cuanto que el ERM mejoró un 63% y eliminó el sesgo sistemático en la predicción de las concentraciones plasmáticas de doxorubicina. Respecto al primer ciclo de quimioterapia, el RECM mejoró un 41% en el segundo ciclo, circunstancia que indica una mejora significativa en la precisión de la predicción de las concentraciones plasmáticas de doxorubicina.
Capacidad predictiva del modelo final en primer y segundo ciclo de quimioterapia
| Exactitud (%) | Precisión (%) | |||||
| Estimación | Bootstrap | Estimación | Bootstrap | |||
| Media | IC 95% | Media | IC 95% | |||
| Ciclo 1 | 27,9 | 28,1 | 12,1-48,3 | 104 | 105 | 76,8-165 |
| Ciclo 2 | 10,4 | 10,5 | −2,1-25,1 | 62,4 | 62,4 | 48,9-78,1 |
IC: intervalo de confianza.
El objetivo de este trabajo ha sido desarrollar un modelo farmacocinético poblacional para caracterizar la evolución temporal de las concentraciones plasmáticas de doxorubicina en pacientes oncológicos. Así, se ha determinado que la farmacocinética de doxorubicina se describe adecuadamente mediante un modelo tricompartimental con distribución lineal a los compartimentos periféricos y eliminación lineal desde el compartimento central. El aclaramiento plasmático poblacional estimado ha sido de 58,8 l/h. Las variabilidades inter e intraindividuales son ligeramente superiores a valores previamente publicados6,7. La variabilidad interindividual es superior a la intraindividual, circunstancia que justifica el potencial valor añadido de la personalización farmacoterapéutica, a la vez que cuantifica la magnitud de la variabilidad en las concentraciones plasmáticas que no se explica por diferencias intraindividuales o por otros errores (toma de muestra, dosis, técnica analítica). El valor típico del volumen de distribución en compartimento central ha sido de 23,8 l, valor comparable al determinado por Callies et al.6 (21,2l) y superior al estimado por Freyer et al.7 (9,3l). El volumen de distribución en estado estacionario estimado ha sido de 2.294 l que claramente excede el volumen de agua corporal total, y refleja la elevada distribución a los tejidos periféricos y concuerda con la elevada afinidad de doxorubicina por su target intracelular, el ADN.
Los resultados de la validación interna del modelo farmacocinético seleccionado han confirmado que el modelo predice adecuadamente la evolución temporal de las concentraciones de doxorubicina y su variabilidad y, por lo tanto, puede utilizarse para la optimización farmacoterapéutica de los tratamientos oncológicos con doxorubicina.
El análisis exploratorio gráfico del efecto de la edad, sexo, superficie corporal, ALT, AST, bilirrubina total, fosfatasa alcalina y tipo de tumor no indican correlación alguna entre las covariables y los parámetros farmacocinéticos. Por lo tanto, y dado el escaso número de sujetos incluidos en el presente estudio, no se ha realizado un análisis formal de los efectos de las covariables en los parámetros farmacocinéticos, aspecto que se evaluará posteriormente cuando el número de pacientes monitorizados sea mayor y se disponga de suficiente poder estadístico para evaluar estas correlaciones.
Para evaluar el efecto que la optimización farmacoterapéutica puede tener en la individualización de pautas posológicas de doxorubicina, se ha determinado la capacidad predictiva del modelo seleccionado en términos de exactitud y precisión. Los resultados indican que en el primer ciclo el modelo presenta un sesgo en la predicción de las concentraciones plasmáticas dado que tienen más peso las estimaciones de la población a priori respecto a la población de estudio. Sin embargo, en el segundo ciclo, al introducir los datos de concentraciones plasmáticas del paciente determinadas tras el primer ciclo de quimioterapia, el sesgo desaparece y la precisión aumenta. La evaluación del número máximo de ciclos de quimioterapia que sería necesario monitorizar se tiene que explorar con más profundidad y será objeto de un futuro trabajo.
En conclusión, el modelo bayesiano poblacional caracteriza adecuadamente la evolución temporal de las concentraciones plasmáticas de doxorubicina en la población de estudio y puede utilizarse de forma exacta y precisa para optimizar las pautas posológicas de doxorubicina en pacientes oncológicos.
Conflicto de interesesLos autores declaran que no hay conflictos de intereses.

