

Dabigatran es el primer anticoagulante inhibidor directo de la trombina disponible por vía oral. La absorción del profármaco dabigatran etexilato y su conversión a dabigatran es rápida (concentraciones máximas de 4-6 h tras cirugía y 2 h posteriormente) y, pese a la baja biodisponibilidad oral, presenta escasa variabilidad entre individuos. Inhibe específica y reversiblemente la trombina, la enzima llave de la cascada de la coagulación. Los estudios tanto en voluntarios sanos como en pacientes sometidos a cirugía ortopédica mayor muestran un perfil pk/pd predecible, lo que permite regímenes fijos de dosificación. El efecto anticoagulante se correlaciona bien con las concentraciones plasmáticas del fármaco, lo que aúna una efectiva anticoagulación con un bajo riesgo de hemorragia. La excreción es mayoritariamente renal (lo que condiciona su dosificación en pacientes ancianos y con insuficiencia renal), y no sufre metabolismo hepático por el sistema del citocromo P450, por lo que presenta un perfil de fármaco sin grandes problemas de interacción con otros medicamentos.
Rivaroxaban será probablemente el primer fármaco anticoagulante oral inhibidor directo del factor Xa (FXa) disponible. Produce una inhibición reversible y predecible de la actividad del FXa con capacidad de inhibir el FXa ligado al coágulo. Sus características farmacocinéticas incluyen rápida absorción, con elevadas biodisponibilidad y unión a proteínas plasmáticas y semivida de eliminación de, aproximadamente, 8 h. La eliminación es de tipo dual, renal (mayoritaria) y biliar. Aunque ha demostrado tener un potencial moderado de interacción con inhibidores fuertes del citocromo P450-A4, no parece inhibir ni inducir ninguna enzima P450.
Dabigatran is the first available oral direct thrombin inhibitor anticoagulant. Absorption of the prodrug, dabigatran etexilate and its conversion to dabigatran is rapid (peak plasma concentrations are reached 4-6 hours following surgery, and a further 2 hours later). Its oral bioavailability is low, but shows reduced interindividual variability. Dabigatran specifically and reversibly inhibits thrombin, the key enzyme in the coagulation cascade. Studies both in healthy volunteers and in patients undergoing major orthopaedic surgery show a predictable pk/pd profile that allows for fixed-dose regimens. The anticoagulant effect correlates adequately with the plasma concentrations of the drug, demonstrating effective anticoagulation combined with a low risk of bleeding. Dabigatran is mainly eliminated by renal excretion (a fact which affects the dosage in elderly and in moderate-severe renal failure patients), and no hepatic metabolism by cytochrome P450 isoenzymes has been observed, showing a good interaction profile.
Rivaroxaban will probably be the first available oral factor Xa (FXa) direct inhibitor anticoagulant drug. It produces a reversible and predictable inhibition of FXa activity with potential to inhibit clot-bound FXa. Its pharmacokinetic characteristics include rapid absorption, high oral availability, high plasma protein binding and a half-life of aprox. 8 hours. Rivaroxaban elimination is mainly renal, but also through faecal matter and by hepatic metabolism. Although the drug has demonstrated moderate potential to interact with strong CYP3A4 inhibitors, it does not inhibit or induce any major CYP450 enzyme.
Actualmente hay varios agentes antitrombóticos utilizados en la práctica clínica, como la heparina no fraccionada (HNF), y agentes relacionados, como las heparinas de bajo peso molecular (HBPM) y el derivado pentasacárido de síntesis fondaparinux, los anticoagulantes orales (como la warfarina y el acenocumarol), los inhibidores directos parenterales de la trombina (lepirudina y bivalirudina) o incluso el ácido acetilsalicílico (aspirina) que han demostrado eficacia y seguridad en su uso1,2.
Sin embargo, estos tratamientos presentan diferentes inconvenientes ya que, por ejemplo, en el caso de la heparina, ésta debe ser administrada por vía parenteral y con la posibilidad de ocasionar trombocitopenia (HIT) además de requerir una estricta monitorización de laboratorio (tiempo de tromboplastina parcial activada [TPPa]). En el caso de las HBPM y el fondaparinux, su principal inconveniente radica en que, además de administrarse por vía parenteral, deben usarse con precaución en insuficiencia renal, además de no disponer de un antídoto que pueda neutralizar eficazmente su actividad en caso de hemorragia. En el caso de los anticoagulantes orales dicumarínicos, los inconvenientes son la necesidad de ajustar las dosis y los controles periódicos de laboratorio para adecuar los valores de la razón internacional normalizada (INR), además de tener en cuenta que el tratamiento puede causar múltiples interacciones con otros fármacos y con algunos alimentos3-5. Pese al gran esfuerzo asistencial en la protocolización y el uso adecuado de los fármacos anticoagulantes disponibles, los resultados en la práctica clínica no son en absoluto los deseados6.
Estas limitaciones han orientado la investigación hacia la búsqueda de fármacos que, administrados por vía oral, inhiban directamente etapas muy definidas de la coagulación y, de ese modo, disminuyan la generación de trombina o, directamente, inhiban el producto final enzimático, la trombina. Esta acción antitrombínica tendría, además, la posibilidad de modificar la activación plaquetaria mediada por trombina, una acción que no poseen los fármacos que inhiben específicamente la función plaquetaria, al menos en el grado necesario para obtener un grado de prevención suficiente de la enfermedad tromboembólica arterial7,8.
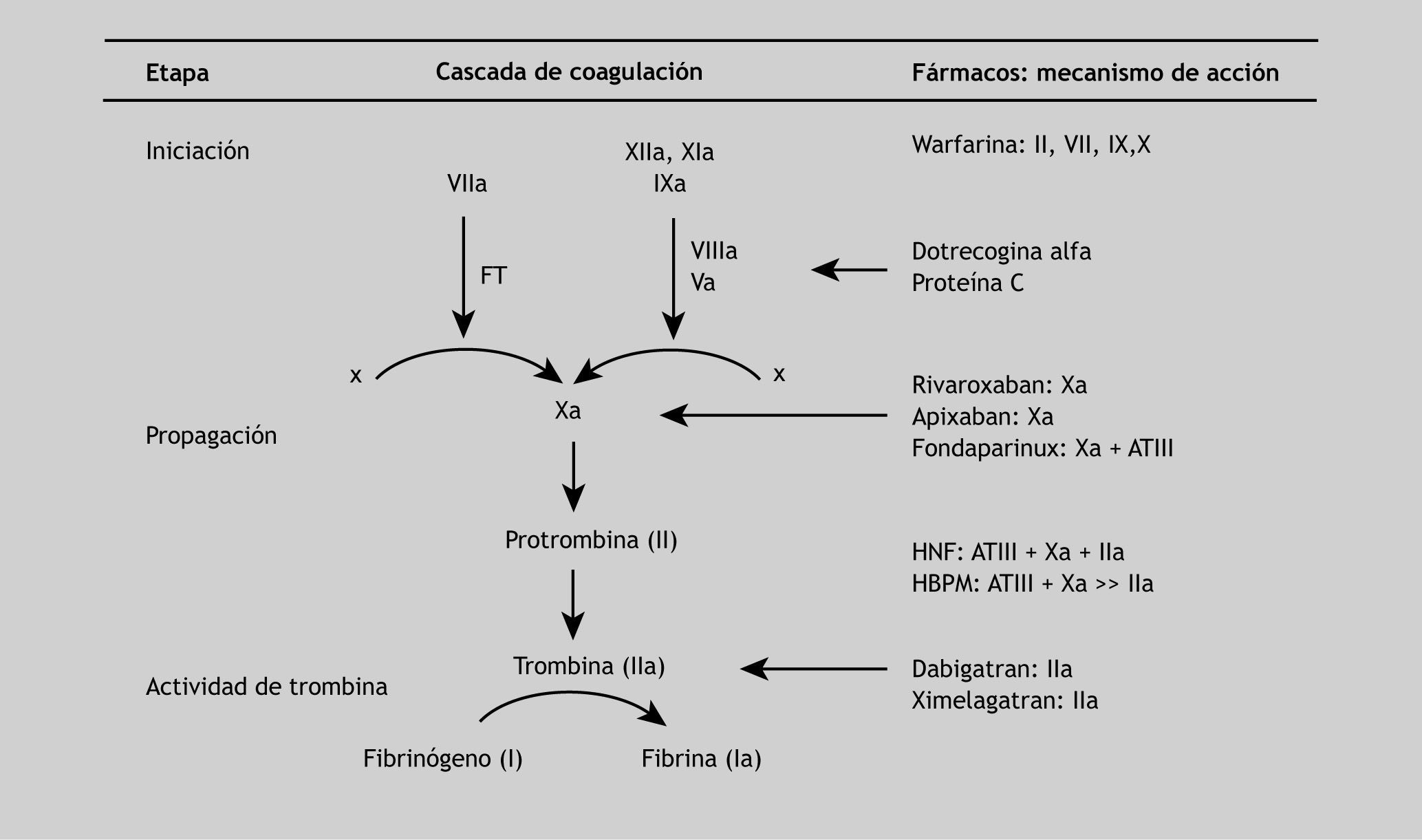
Tras una época en la que apenas ha habido innovaciones farmacológicas en el campo de los anticoagulantes, empiezan a surgir nuevos fármacos que deberán demostrar que son más eficaces y seguros que los tradicionales. En este sentido, se han investigado nuevos anticoagulantes orales que actúan mediante diferentes mecanismos de acción, como los inhibidores directos de la trombina y los inhibidores directos del factor Xa9 (fig. 1).

En España, se ha comercializado recientemente un nuevo inhibidor directo de la trombina oral, el dabigatran etexilato Pradaxa® (Boehringer Ingelheim), para la prevención de la tromboembolia venosa (TEV) en adultos sometidos a cirugía de reemplazo de cadera o rodilla. Asimismo, la Agencia Española de Medicamentos y Productos Sanitarios ha autorizado un nuevo fármaco inhibidor directo del factor Xa oral, rivaroxaban (Xarelto®), desarrollado por Bayer HealthCare y Johnson & Johnson Pharmaceutical Research & Development, para la prevención de formación de coágulos sanguíneos venosos en pacientes sometidos a cirugía electiva de sustitución total de cadera y rodilla7,8.
El objetivo principal de esta revisión es evaluar las propiedades farmacodinámicas y farmacocinéticas de los fármacos dabigatran etexilato y rivaroxaban, dos medicamentos que, según algunos expertos, tienen el potencial de cambiar los estándares de la práctica clínica en la prevención de la trombosis venosa profunda y la embolia pulmonar.
Propiedades químicas y farmacodinámicasCaracterísticas y origenEl reciente desarrollo de nuevos anticoagulantes orales con mecanismos de acción novedosos sobre dianas terapéuticas en la coagulación parece que podría cambiar los estándares actuales en la farmacoterapia anticoagulante10,11. Entre los inhibidores directos orales de la trombina el primero comercializado en España ha sido dabigatran (DB). Por otra parte rivaroxaban, el primer fármaco anticoagulante oral inhibidor del factor Xa ha sido recientemente autorizado por la EMEA. Tanto la fórmula molecular de dabigatran (C35H45-N7O8S) como de rivaroxaban (C19H18ClN3O5S) corresponden a pequeñas moléculas no peptídicas con pesos moleculares de 628 Da y 436 Da respectivamente. Esto supone una ventaja en la absorción frente a anticoagulantes con mecanismos de acción similares pero de estructura peptídica y elevado peso molecular (heparina), así como HBPM.
La investigación sobre los derivados de la bencidamina (α-NAPAP) por su interés terapéutico como potentes inhibidores de serinproteasas, como la trombina y la tripsina, condujo al descubrimiento de DB que se desarrolló con un perfil farmacocinético mejorado. La adición de una cadena lateral hidrófuga a DB permite la absorción oral del profármaco, dabigatran etexilato (DBE), que requiere de hidrólisis para ser activado12,13. El descubrimiento de rivaroxaban se debe, en cambio, a los cribados farmacológicos de derivados de oxazolidinona, todos ellos potentes inhibidores directos del factor Xa de la coagulación14,15. El estudio de estos derivados permitió desarrollar moléculas con características farmacocinéticas adecuadas, como se ha demostrado en estudios preclínicos16. Es el caso de rivaroxaban, que presenta una elevada biodisponibilidad en parte debida a una característica fisicoquímica única, la ausencia de un grupo altamente básico en su dominio de unión al sitio activo de la enzima15. Apixaban es otro fármaco inhibidor directo del factor Xa que se encuentra actualmente en fase III de investigación clínica17.
Ambos fármacos, dabigatran (Pradaxa®) y rivaroxaban (Xarelto®) han sido autorizados con la indicación de prevención primaria de episodios tromboembólicos venosos en pacientes adultos sometidos a cirugía electiva de reemplazo total de cadera o rodilla. Pradaxa® se comercializa en cápsulas duras de 75 y 110 mg que contienen múltiples pildoritas de un diámetro aproximado de 0,8 mm. El profármaco DBE se encuentra recubriendo las pildoritas que contienen un núcleo de ácido tartárico. Este núcleo proporciona un microentorno ácido que facilita la absorción de DBE independientemente de las variaciones del pH gástrico18,19. En cuanto a Xarelto® está próxima su comercialización en España en forma de comprimidos de 10 mg con recubrimiento pelicular. Los comprimidos son de liberación inmediata y se formulan a partir de rivaroxaban en polvo micronizado para facilitar su disolución, puesto que se trata de una sustancia de clase II según el Sistema de Clasificación Biofarmacéutica (baja solubilidad, alta permeabilidad)20.
Mecanismos de acciónLa trombina (factor IIa) es el efector final de la cascada de la coagulación que cataliza la formación de fibrina a partir de fibrinógeno plasmático. Además de su papel en la coagulación, se trata del agonista fisiológico más potente de la activación plaquetaria, por ello representa una diana terapéutica clave en el desarrollo de los nuevos fármacos anticoagulantes orales. Por otra parte, el factor Xa actúa como punto de convergencia de las vías intrínseca y extrínseca de la coagulación y cataliza la conversión de protrombina a trombina. Una sola molécula del factor Xa puede generar más de 1.000 de trombina21,22; en consecuencia la inhibición del factor Xa puede bloquear este proceso al disminuir la activación de la coagulación y de las plaquetas mediadas por trombina16,23.
Tanto si la cascada de la coagulación es inhibida a nivel de la trombina como del factor Xa, o incluso más arriba en la secuencia, el resultado neto es una disminución de la actividad de la trombina. Por el momento, los ensayos clínicos con inhibidores directos de la trombina (IDT) e inhibidores orales del factor Xa siguen caminos paralelos, por lo que la respuesta a cuál es el grupo de fármacos con mayores ventajas se resolverá con el diseño de ensayos clínicos comparativos24.
Dabigatran es un potente IDT, competitivo y reversible. Al igual que melagatran se une exclusivamente al sitio activo o catalítico de la trombina y causa su inactivación, por ello se considera que ambos IDT son univalentes25. La inhibición de la trombina es dependiente de la concentración y ésta se produce tanto en la trombina unida a fibrina como en la trombina libre. Por otra parte, la elevada selectividad de DB por trombina (también tripsina, aunque ésta sólo es activa en el intestino delgado) y su unión reversible con la trombina le confieren un perfil más seguro y predecible que el de las hirudinas (IDT parenterales), que presentan unión irreversible (no covalente).
A diferencia de DB, rivaroxaban no inhibe la trombina (factor II activado) y no se ha observado que produzca efectos en las plaquetas. Rivaroxaban, además de inhibir el factor Xa libre, produce la inhibición in vitro del factor Xa unido al complejo de la protrombinasa, lo que permite suponer que podría inhibir el factor Xa unido al coágulo, a diferencia de las heparinas de bajo peso molecular y fondaparinux26,27.
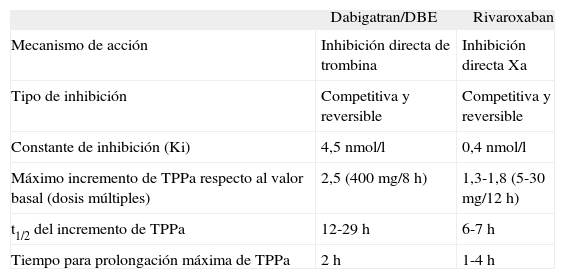
Una característica común a los mecanismos de acción de DB y rivaroxaban es que ambos actúan sobre sus respectivas dianas de forma directa, al igual que sucede con los IDT parenterales (hirudina) y en contraposición al mecanismo de acción indirecto de la heparina, que actúa como cofactor de la antitrombina III, que es el inhibidor fisiológico de la trombina. En la tabla 1 se resumen estas características farmacológicas.
Principales características farmacodinámicas de dabigatran, dabigatran etexilato (DBE) y rivaroxaban
| Dabigatran/DBE | Rivaroxaban | |
| Mecanismo de acción | Inhibición directa de trombina | Inhibición directa Xa |
| Tipo de inhibición | Competitiva y reversible | Competitiva y reversible |
| Constante de inhibición (Ki) | 4,5 nmol/l | 0,4 nmol/l |
| Máximo incremento de TPPa respecto al valor basal (dosis múltiples) | 2,5 (400 mg/8 h) | 1,3-1,8 (5-30 mg/12 h) |
| t1/2 del incremento de TPPa | 12-29 h | 6-7 h |
| Tiempo para prolongación máxima de TPPa | 2 h | 1-4 h |
TPPa: tiempo de tromboplastina parcial activada.
Dabigatran se comporta como un inhibidor potente, competitivo y reversible de la trombina humana, así como de la agregación plaquetaria inducida por trombina. Su constante de inhibición (Ki) es de 4,5 nmol/l y la concentración necesaria para inhibir el 50 % de la trombina generada (IC50) es 0,56 μmol/l; su capacidad de inhibición es similar a la ya mostrada por melagatran en estudios in vitro28. La inhibición en la actividad de la trombina y, en consecuencia, en la formación de trombos resultó ser dependiente de la dosis y alcanza la inhibición máxima tras 1 h de la administración oral. Asimismo, se demostró que tras este rápido inicio del efecto, éste disminuía en paralelo con la eliminación de DB; se apreció una disminución rápida del efecto seguida de una fase lenta terminal19. Los estudios preclínicos con DB en ratas y monos rhesus demostraron su elevada potencia anticoagulante administrado tanto vía intravenosa como vía oral (en forma de DBE)29.
Rivaroxaban inhibe el factor Xa también de forma competitiva y reversible al unirse a su centro activo con una IC50 de 21 nmol/l. La inhibición del factor Xa (sobre el cual tiene una especificidad mucho mayor que para otras serinproteasas) también es dependiente de la dosis y en estudios preclínicos el grado máximo de inhibición (Emáx) osciló entre el 20 y el 61 % para el intervalo de dosis de 5-80 mg, que se alcanza 1 a 4 h después de la administración oral de dosis únicas de rivaroxaban30.
En cuanto a los efectos de DB en los parámetros de coagulación, se apreció una prolongación de los tiempos cuando se evaluó tanto sobre sujetos sanos como pacientes intervenidos de cirugía ortopédica de cadera (COC)31. En el ensayo BISTRO I en pacientes sometidos a COC se puso en evidencia que el tiempo de tromboplastina parcial activada (TPPa) se incrementa de forma no proporcional a las concentraciones de DB, tal y como sucede en sujetos sanos32. En consecuencia, el TPPa presenta una relación curvilínea que alcanza una meseta a concentraciones superiores a 400 ng/ml; por este motivo no resulta un buen parámetro para cuantificar el efecto anticoagulante de DB, especialmente a concentraciones plasmáticas altas.
Por otra parte, se observó una relación lineal entre las concentraciones de DB y el incremento del tiempo de coagulación de la ecarina (ECT), aunque se debe tener en cuenta que ECT tiene una mayor sensibilidad justo al iniciar el tratamiento con DB inmediatamente después de la cirugía, por lo que es necesaria una reducción del 50 % de la dosis el día de la cirugía. En este sentido, el ECT o, mejor aún, el ensayo cromogénico de ecarina es el más adecuado en términos de sensibilidad y precisión para evaluar el efecto anticoagulante de DB, así como de otros IDT, como las hirudinas19,33. No obstante, en la práctica clínica no se recomienda la monitorización de parámetros de coagulación, sino la monitorización clínica de los pacientes con mayor riesgo de hemorragia.
En los ensayos clínicos en fase I y II, los efectos anticoagulantes de rivaroxaban se evaluaron midiendo la inhibición del factor Xa, así como la prolongación del tiempo de protrombina (TP) y TPPa. Puesto que los inhibidores del factor Xa tienen su efecto en el punto de convergencia de las vías intrínseca y extrínseca de la coagulación, es de esperar que afecten tanto el TP como el TPPa. De hecho, rivaroxaban prolonga tanto el TP como el TPPa de forma dependiente de la dosis, si bien el TP puede ser más sensible a rivaroxaban. En cambio éste no tiene ningún efecto en el ECT, debido a su alta especificidad16,30. En la práctica clínica, y al igual que para DB, no es necesario monitorizar los parámetros de coagulación durante el tratamiento con rivaroxaban gracias a sus predecibles farmacocinética y farmacodinamia. Otra ventaja observada frente a otros anticoagulantes es que rivaroxaban no presenta reacción cruzada con los anticuerpos que causan la trombocitopenia inducida por heparina. En la actualidad, siguen en marcha diversos ensayos clínicos en fase III para la evaluación de rivaroxaban en prevención de TEV en cirugía ortopédica (ensayos RECORD), así como para otras indicaciones, como prevención de ictus en fibrilación auricular (ensayo ROCKET AF) y prevención secundaria en síndrome coronario agudo (ensayo ATLAS).
Las dosis de DBE (Pradaxa®) aprobadas para la prevención primaria de la TEV en pacientes adultos fueron 110 mg en las primeras 4 h tras la cirugía y una dosis de mantenimiento de 220 mg (2 cápsulas) una vez al día durante 10 días en prótesis de rodilla o 28-35 días en prótesis de cadera. En cuanto a rivaroxaban (Xarelto®), las dosis aprobadas son de 10 mg; la primera dosis en las primeras 6-10 h tras la intervención y luego dosis de mantenimiento de 10 mg al día durante 2 semanas en prótesis de rodilla y 5 semanas en prótesis de cadera.
Propiedades farmacocinéticasAbsorciónRivaroxaban es un compuesto no básico que es rápidamente absorbido y presenta una biodisponibilidad del 60-80 % tras su administración oral30,34. Un reciente análisis farmacocinético poblacional de rivaroxaban, que utilizaba los datos de dos ensayos clínicos de fase II35, muestra que la absorción oral es rápida, con un tiempo máximo (tmáx) de 1-2 h. En estado estacionario, y administrado junto con la comida, el tmáx se retrasa a 2,5-3 h23. En comparación con la cinética en ayunas, al tomarlo con la comida se produce un incremento en el Cmáx y en el AUC cercano al 30 %, mientras el tmáx se retrasa significativamente (de 2,75 h en ayunas hasta 4 h con alimentos), si bien también se observa una reducción en la variabilidad farmacocinética. Además, estas diferencias farmacocinéticas se trasladan a los valores farmacodinámicos; la administración en ayunas reduce ligeramente el valor máximo del tiempo de protrombina y la máxima inhibición de la actividad del factor Xa. Estos datos han conducido a que rivaroxaban se administre junto con la comida en todos los ensayos clínicos23. La administración de ranitidina o antiácidos no afecta significativamente a su absorción en sujetos sanos36.
DBE se absorbe en el estómago y el intestino delgado y, una vez absorbido, DBE se convierte en su metabolito activo, DB, mediante una reacción de hidrólisis catalizada por una esterasa. Esta reacción se da principalmente en el enterocito, la vena porta y el hígado, donde se producen dos metabolitos intermedios, BIBR 951 y BIBR 1087, de forma inmediata y resulta especialmente difícil detectar las concentraciones del DBE o de los dos metabolitos intermedios en el plasma de sujetos sanos, en los que los AUC de estos sustratos, comparados con el de DB, son < 0,4 %24. La biodisponibilidad de DBE es baja, en individuos sanos se encuentra entre el 5 y el 7 %, y el tiempo necesario para alcanzar la concentración máxima de fármaco en sangre está entre 0,5 y 2 h37.
DBE ha sido bien tolerado en población joven (18-45 años) y en población de edad más avanzada (65-87 años), tanto en dosis única diaria de 10 a 400 mg como en dosis múltiples diarias de 50-400 mg dos/tres veces al día. Estudios recientes han demostrado que tanto DBE en solución como DBE en forma de comprimidos peliculares (ambas formas farmacéuticas formuladas junto con ácido tartárico) siguen una cinética de primer orden en el proceso de absorción, en dosis que van desde 10 a 1.200 mg/día: incrementos en la dosis se corresponden con incrementos proporcionales en la Cmáx y en el AUC. Además, se puede descartar la presencia de efecto de primer paso saturable aun a dosis de 1.200 mg/día19.
La administración de DBE junto con alimentos no modifica la biodisponibilidad en magnitud. Dos estudios en voluntarios sanos (n = 12 y n = 18), en los que se administraba una dosis de fármaco de 150 mg, junto con alimentos lipídicos y altamente calóricos, concluyeron que el tiempo necesario en alcanzar la Cmáx se retrasaba 2 h respecto a la toma del fármaco en ayunas19, mientras que la biodisponibilidad en magnitud permanecía igual. Además, el retraso en la absorción disminuye la variabilidad interindividual de Cmáx del 42 al 24 % y de AUC del 44 al 21 %, con lo que resultan más predecibles las concentraciones plasmáticas del fármaco19,31.
La administración concomitante de pantoprazol y DBE produce una disminución del 22 % del AUC de DB (desde 904 a 705 ng/h/ml) y del 33 % de la Cmáx (desde 111 a 705 ng/ml). Sin embargo, a las dosis recomendadas de 150 mg y 220 mg, la curva dosis-respuesta de dabigatran se encuentra en un estado bastante aplanado, por lo que reducciones en AUC y Cmáx producidas por el inhibidor de la bomba de protones no se consideran clínicamente relevantes.
Pacientes convalecientes de una cirugía mayor presentan una variabilidad farmacocinética entre individuos de DB mucho más elevada (el 69 % para el AUC y el 65 % para la Cmáx) que la que presentan individuos sanos19,31. En pacientes sometidos a COC, tanto la velocidad como el grado de absorción de DBG se ven reducidos en las primeras 24 h tras la intervención, la Cmáx se alcanza a las 6 h, aunque el AUC no se modifica31. La variabilidad entre individuos resulta ser mayor durante el primer día tras la intervención, en comparación con los sucesivos días, como se demostró en un estudio farmacocinético (n = 287), en el que se observaron diferentes perfiles de absorción. Éstos pueden deberse a efectos de la cirugía, enlentecimiento de la motilidad gastrointestinal y cambios en el pH gástrico, algunos posiblemente consecuencia de la anestesia, aunque en este tipo de pacientes el perfil farmacocinético de DBE no fue alterado por los opioides. Esta variación estaba influida por la edad y la creatinina sérica38.
En este tipo de pacientes quirúrgicos, también rivaroxaban tiene un perfil de absorción adecuado y un incremento en la dosis produce un incremento proporcional en su concentración plasmática. En ambos fármacos, en mayor o menor medida, la absorción se ve enlentecida y tiene como resultado una Cmáx no tan elevada, que podría reducir el riesgo de hemorragia aun cuando la administración del fármaco fuera poco después de la intervención.
DistribuciónDB es un fármaco bicompartimental, que sigue una cinética de distribución de primer orden, en la que el volumen aparente de distribución, el aclaramiento plasmático y la semivida de eliminación son independientes de la dosis administrada. Mediante el método de Wagner, una dosis única de DBE fue capaz de predecir el perfil farmacocinético en estado estacionario39. En las primeras 4-6 h tras la administración oral, la fase de disposición rápida hace que la concentración plasmática de DB disminuya más del 70 %, seguidamente se produce una fase de disposición lenta en la que la concentración se encuentra más mantenida. La t1/2 es de 12 a 14 h, tanto en jóvenes sanos como en los de más edad y algo más (12-17 h) en pacientes convalecientes de cirugía ortopédica mayor. Se requiere de 2-3 días en sujetos jóvenes sanos –hasta 7 días en pacientes con fibrilación auricular– para alcanzar el estado estacionario, en régimen de dosis múltiples39. DB no se une de manera significativa a proteínas plasmáticas (un 30 %, aproximadamente). Su volumen de distribución aparente es de 60-70 l, lo que indica una moderada distribución a los tejidos37. Las estimaciones para los compartimentos central y periférico fueron 30,8 l (error estándar [EE] del 17 %) y 136 l (EE del 42 %), respectivamente, según análisis farmacocinéticos poblacionales en pacientes sometidos a cirugía de cadera37. La variabilidad interindividual de la farmacocinética de DB también se incrementa, prácticamente el doble (coeficiente de variación del 60 %) en este tipo de pacientes.
Rivaroxaban muestra una farmacocinética y una farmacodinámica proporcionales a las dosis utilizadas, con una semivida de eliminación terminal media de 5 a 9 h, sin evidencias de acumulación en el estado estacionario con ninguna de las dosis ensayadas23,35. Por el contrario, se une a proteínas plasmáticas en más de un 90 %30,34, muestra un volumen de distribución aparente aproximado de 50 l, posee una afinidad moderada por proteínas tisulares y no se acumula en órganos ni tejidos.
Ninguno de los dos fármacos tiende a acumularse en el organismo y tampoco necesitan de ajuste posológico por peso corporal. En todo caso, la experiencia clínica de DBE se limita a pacientes con pesos de 50-110 kg. En el caso de rivaroxaban, pacientes de hasta 120 kg no sufrieron ningún cambio significativo en su Cmáx en la dosis establecida, pero en pacientes con peso menor de 50 kg, la Cmáx aumentó un 24 %. Aun así, la recomendación es utilizar dosis fijas y seguir más de cerca a estos pacientes.
EliminaciónRivaroxaban se excreta mediante un proceso dual, por vía renal (66 %) y biliar (28 %). Un 36 % del fármaco se excreta de forma inalterada por la orina; otra vía de eliminación incluye el metabolismo hepático vía citocromo P450-A423,30. En sujetos jóvenes, rivaroxaban presenta una semivida de eliminación de unas 9 h, que puede incrementarse hasta 12 h en pacientes ancianos, en pacientes con insuficiencia renal y posquirúrgicos.
El aclaramiento plasmático de rivaroxaban se correlaciona con el aclaramiento de creatinina (ClCr). El AUC del fármaco se incrementa en un 44, un 52 y una 64 % en pacientes con insuficiencia renal leve (ClCr, 30-49 ml/min), moderada (30-49 ml/min) o grave (< 30 ml/min) en comparación con el grupo control de pacientes con función renal normal40. Esta elevación en el AUC también se traslada al efecto farmacodinámico: la actividad anti-Xa, el TP y el TPPa se incrementan de forma muy significativa.
En pacientes sometidos a COC, la variabilidad interindividual en el aclaramiento de rivaroxaban en los primeros 3 días tras la cirugía (coeficiente de variación del 70 %) resulta ser significativamente mayor que la observada en días posteriores (39 %). Esta diferencia, habitual en medicamentos orales tras la cirugía, puede responder a efectos en los flujos renal y hepático tras cirugía mayor35. Los pacientes intervenidos de COC presentan un aclaramiento plasmático mayor (un 26 % mayor) que los intervenidos de prótesis de rodilla, por motivos no identificados en el análisis farmacocinético poblacional35.
Estudios de fase I demuestran que el sexo y el peso corporal no influyen significativamente en las propiedades farmacológicas de rivaroxaban, de ahí que se recomienden dosis fijas diarias en una amplia serie de pacientes: edad (intervalo, 18-94 años), sexo, peso corporal (intervalo, 37-173 kg), insuficiencia renal leve a moderada e insuficiencia hepática leve23,34. En cualquier caso, debe tenerse presente que se ha excluido a los pacientes con insuficiencia renal y hepática grave de los ensayos clínicos disponibles.
Estudios in vitro indican que rivaroxaban presenta un potencial moderado de interacción con inhibidores potentes del citocromo P450-A4, aunque por sí mismo no parece inducir ni inhibir las principales enzimas del citocromo P45041.
DB no es metabolizado por citocromos hepáticos ni por enzimas oxidorreductasas, ni siquiera en la primera reacción que sufre su profármaco37. Estudios in vitro en microsomas hepáticos indican que la reacción sufrida por DBE a sus intermediarios BIBR 1087 y a DB es catalizada por carboxilesterasas microsomales. Por lo que no presenta ninguna interacción con inhibidores o inductores del metabolismo hepático ni ha visto alterado su perfil farmacocinético en sujetos jóvenes con daño hepático moderado (Child-Pugh clase B). La eliminación de DB se produce mayoritariamente por la orina de forma inalterada (en un 85 %), con tasa de filtración glomerular de 100 ml/min. El remanente es conjugado con ácido glucurónico por una reacción catalizada por las acilglucuronidasas y es eliminado vía biliar/fecal (6 %). Estos conjugados son farmacológicamente activos, con 4 isómeros de actividad similar a DB no conjugado y se pueden encontrar en cantidades muy pequeñas en la orina37,39.
Debido a que la eliminación de DB es principalmente renal, en pacientes con una función renal deteriorada (ClCr < 50 ml/min), disminuye la velocidad de eliminación y se obtienen concentraciones elevadas de DB, por lo que puede plantearse una reducción de dosis. El primer día tras la intervención estos pacientes presentan un aclaramiento plasmático aparente de DB un 30 % menor38, y la semivida de eliminación para este subgrupo poblacional es de 14-17 h.
En pacientes con insuficiencia renal moderada (ClCr = 30-50 ml/min) el AUC se incrementa 2,7 veces, por lo que se recomienda utilizar una dosis de 150 mg/día y la mitad de ésta (75 mg), el día antes de la operación. La administración de DBE está contraindicada en pacientes con insuficiencia renal grave (ClCr < 30 ml/min), donde el AUC se ve aumentada 6 veces y la t1/2 es el doble19. Aunque algunos estudios han propuesto el uso de dosis fijas de DBE para la mayoría de los pacientes con insuficiencia renal leve, moderada y grave, en la población anciana (> 75 años), puesto que generalmente su función renal está deteriorada, se recomienda una dosis de 150 mg diaria. En estudios realizados en población anciana, se observó que a igual dosis, éstos presentaban concentraciones plasmáticas 1,8 veces mayores y valores de AUC y Cmáx incrementados un 50 y un 25 %, respectivamente, en comparación con voluntarios jóvenes19.
En pacientes sometidos a cirugía ortopédica de cadera, el perfil farmacocinético de DBE no se altera por diferencias de sexo, consumo de alcohol o tabaco, aunque pacientes de edad avanzada y tras cirugía sí que pueden ver disminuida la eliminación del fármaco ya que su ClCr suele ser menor, por lo que en éstos y en pacientes de peso corporal < 50 o > 110 kg, se recomienda un estrecha monitorización clínica. En niños y adolescentes no hay suficiente experiencia, por lo que su uso en éstos no estaría aconsejado. El valor de ALT debe determinarse como parte de la evaluación preoperatoria estándar. De hecho, se excluyó de los ensayos clínicos a los pacientes con elevación de las enzimas hepáticas más de 2 veces el límite superior de la normalidad (LSN), por lo que tampoco se recomienda su uso en éstos. El sexo y el origen étnico tampoco afectan de un modo clínicamente relevante a la farmacocinética del fármaco.
Estudios en animales muestran toxicidad reproductiva para ambos fármacos por lo que no se recomiendan en embarazo y, dada la ausencia de datos, tampoco durante la lactancia materna18. Tampoco hay experiencia en niños por lo que no deberían indicarse en esta población.
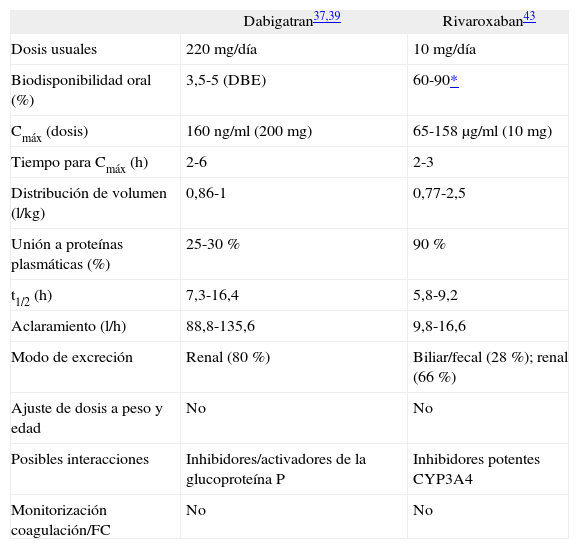
En la tabla 2 se resume la comparación de las características farmacocinéticas de los dos medicamentos.
Principales características farmacocinéticas de dabigatran, dabigatran etexilato (DBE) y rivaroxaban en pacientes tipo
| Dabigatran37,39 | Rivaroxaban43 | |
| Dosis usuales | 220 mg/día | 10 mg/día |
| Biodisponibilidad oral (%) | 3,5-5 (DBE) | 60-90* |
| Cmáx (dosis) | 160 ng/ml (200 mg) | 65-158 μg/ml (10 mg) |
| Tiempo para Cmáx (h) | 2-6 | 2-3 |
| Distribución de volumen (l/kg) | 0,86-1 | 0,77-2,5 |
| Unión a proteínas plasmáticas (%) | 25-30 % | 90 % |
| t1/2 (h) | 7,3-16,4 | 5,8-9,2 |
| Aclaramiento (l/h) | 88,8-135,6 | 9,8-16,6 |
| Modo de excreción | Renal (80 %) | Biliar/fecal (28 %); renal (66 %) |
| Ajuste de dosis a peso y edad | No | No |
| Posibles interacciones | Inhibidores/activadores de la glucoproteína P | Inhibidores potentes CYP3A4 |
| Monitorización coagulación/FC | No | No |
Rivaroxaban no muestra interacciones farmacológicas relevantes con antiinflamatorios no esteroideos (AINE)33,34 o digoxina23,41-43, y aunque se encontraron efectos aditivos con enoxaparina en la actividad anti-Xa (del 48 y el 43 % comparado con rivaroxaban y enoxaparina solos, respectivamente) y del TP (incremento del 38 % comparado con enoxaparina) no fueron relevantes desde el punto de vista clínico23. Estos hallazgos indican que rivaroxaban y enoxaparina pueden administrarse concomitantemente o en terapia secuencial. Rivaroxaban prolonga el tiempo de hemorragia cuando se administra conjuntamente con ácido acetilsalicílico o clopidogrel, si bien la relevancia de esta interacción es escasa por no afectar a la agregación plaquetaria. Dada la elevada unión a proteínas plasmáticas (90 %) de rivaroxaban, es posible que se produzcan interacciones. No presenta reacción cruzada con los sueros de pacientes con HIT, por lo que podría ser una alternativa terapéutica a las heparinas en pacientes con este problema41-43.
DB no se metaboliza por el sistema del citocromo P450 e, in vitro, no tiene efectos en las enzimas del citocromo P450 humano. Sin embargo, la exposición a DB en sujetos sanos aumenta un 60 % en presencia de amiodarona, debido a la inhibición del transportador de la glucoproteína P del que es sustrato DB, por lo que debe reducirse la dosis del anticoagulante a 150 mg/día. Por ello, debe tenerse precaución con el uso de inhibidores del transportador (verapamilo, claritromicina), así como sus inductores (rifampicina, hierba de San Juan) de forma concomitante con DB. No se recomienda el uso concomitante de DB y heparina y derivados, trombolíticos, antagonistas de los receptores GPIIb/IIIa, clopidogrel, ticlopidina y antagonistas de la vitamina K o AINE de larga semivida biológica de eliminación (> 12 h), por el riesgo hemorrágico. Los inhibidores de la bomba de protones disminuyen un 30 % el AUC de dabigatran, pero este efecto carece de relevancia clínica18,19.
Ninguno de los dos fármacos precisa monitorización de pruebas de coagulación en las indicaciones aprobadas pero no hay antídoto específico, por lo que en caso de sobredosificación deberá mantenerse una diuresis adecuada, hemostasia quirúrgica o transfusión de plasma congelado reciente.
ConclusionesLos anticoagulantes orales de nueva generación, dabigatran y rivaroxaban, son los primeros de una nueva serie de antitrombóticos no relacionados con los cumarínicos, que inhiben de forma selectiva y directa la trombina y el factor Xa, respectivamente.
La posibilidad de eliminar los controles de coagulación (si no en todos, sí en la gran mayoría de los pacientes tratados), la administración por vía oral a dosis fijas y una vez al día, con una farmacocinética y una farmacodinámica predecibles en una amplia serie de pacientes y con un perfil de interacciones farmacológicas muy favorable, han servido de base para su desarrollo clínico posterior.
Las características farmacocinéticas y farmacodinámicas de dabigatran etexilato, escasa variabilidad en la absorción por vía oral, rápido inicio de acción, velocidad de eliminación que permite una dosis/día, escasa interacción con alimentos, ausencia de interacciones relevantes y excelente correlación entre concentraciones plasmáticas y efecto farmacológico, brindan un fármaco que, además de ser novedoso en cuanto a su mecanismo de acción, parece de sencillo uso en la práctica clínica.
Rivaroxaban, un nuevo y prometedor anticoagulante oral, presenta en relación con DBE algunas ventajas potenciales, como la menor dependencia de eliminación renal, pero otras características, como su extensa unión a proteínas plasmáticas y su interacción con otros fármacos mediante el sistema del citocromo P450 hepático, deberán ser estudiadas más extensamente.

