

Validación de un método analítico para la determinación de concentraciones de linezolid (LNZ) en fluidos biológicos: plasma, humor vítreo y líquido cefalorraquídeo mediante cromatografía líquida de alta eficacia y posterior detección ultravioleta.
MétodoEl método se validó mediante el estudio de los siguientes parámetros: exactitud, precisión, sensibilidad, linealidad y recuperación. El fármaco se extrajo de la matriz biológica mediante una precipitación proteica con ácido perclórico. La separación cromatográfica se realizó mediante la elución de LNZ con una fase móvil compuesta por el 80% de un tampón de fosfato dipotásico-monohidrogenado (K2HPO4) (15mM; pH=5) con el 20% de acetonitrilo y una fase estacionaria NOVAPAK® C18 150×3,9mm con precolumna. La longitud de onda de lectura fue de 254nm y el flujo de trabajo fue de 1ml/min.
ResultadosSe obtuvieron valores de exactitud entre el 94,4–106,1% y de precisión entre el 0,88–6% y el 3,7–5,6% para la variabilidad intradía e interdía, respectivamente. La recuperación obtenida tras el análisis de las muestras de plasma fue del 93%. El método mostró ser lineal para los intervalos de concentraciones estudiados.
DiscusiónEl método se comporta de forma lineal, precisa y exacta. Además, es rápido, sensible y de bajo coste económico. Es un método útil para la determinación de concentraciones de LNZ en múltiples matrices biológicas. Es posible su utilización como base para posteriores estudios de farmacocinética clínica.
Evaluation of an analytic method for determining linezolid concentrations in biological fluids including plasma, vitreous humour and cerebrospinal fluid using high-efficiency liquid chromatography and subsequent ultraviolet detection.
MethodThe method was validated by studying the following parameters: accuracy, precision, sensitivity, linearity and recovery. The drug was extracted from the biological matrix by means of a protein precipitation with perchloric acid. Chromatographic separation was performed by eluting linezolid with a mobile phase consisting of 80% K2HPO4 buffer solution (15mM; pH=5) and 20% acetonitrile, and a stationary phase, NOVAPAK C18 150×3.9mm with precolumn. The wavelength reading was 254nm and the working flow rate was 1ml/min.
ResultsWe obtained values with accuracies between 94.4 and 106.1%, and precisions between 0.88–6% and 3.7–5.6% for intra-and inter-day variability, respectively. Recovery obtained after analysing the plasma samples was at 93%. The method showed itself to be linear for the concentration levels under study.
DiscussionThe method's behaviour can be described as linear, precise and accurate. Furthermore, the method is fast, sensitive, and inexpensive. It is useful for determining linezolid concentrations in multiple biological matrices. It can also be used as a basis for further clinical pharmacokinetic studies.
El linezolid (LNZ) es una oxizolidinona sintética que actúa inhibiendo la iniciación de la síntesis de proteínas bacterianas1. Tiene un amplio espectro de actividad frente a microorganismos grampositivos: estafilococos resistentes a meticilina, neumococos resistentes a penicilina, Enterococcus faecalis y Enterococcus faecium resistentes a vancomicina2.
Farmacocinéticamente, cabe destacar que LNZ presenta una completa biodisponibilidad oral (100%), una semivida de eliminación de 5–6h y una escasa unión a proteínas plasmáticas (el 31%). Además, presenta un volumen de distribución de 40–50l, lo que indica una buena distribución a los tejidos, alcanzando concentraciones altas en la piel, el líquido sinovial, el humor acuoso, el líquido cefalorraquídeo (LCR), el parénquima pulmonar, etc3. El LNZ es una buena alternativa terapéutica en infecciones de derivaciones ventriculoperitoneales y otras infecciones de cuerpos extraños, en los que predominan los microorganismos grampositivos4. En estos casos es crucial conocer el grado de penetración del antibiótico para asegurar que se alcanzan concentraciones terapéuticas que permitan su empleo en sospecha o confirmación microbiológica de la infección.
La cromatografía es una técnica analítica de separación con gran sensibilidad, selectividad y exactitud. Es idónea para la separación de especies no volátiles, termolábiles, aminoácidos, proteínas, hidrocarburos y fármacos en general, y está considerada una técnica de referencia en los estudios de investigación. En la literatura médica, se encuentran descritos varios métodos cromatográficos para la determinación de LNZ en suero, orina y plasma5–7.
El objetivo principal de este estudio es el desarrollo, puesta a punto y validación de un método analítico rápido, preciso y sencillo para la determinación de LNZ en plasma, LCR y humor vítreo (HV) mediante cromatografía líquida de alta eficacia y detección ultravioleta (UV).
Material y métodosReactivosEl LNZ fue suministrado por Pharmacia & Upjohn (pureza del 100%). El acetonitrilo para cromatografía líquida de alta eficacia, ácido perclórico y el fosfato dipotásico-monohidrogenado (grado análisis p.a.) fueron suministrados por Merck-Farma y Química, S. L. (Barcelona) y el ácido ortofosfórico por Panreac Química S. A. (Barcelona).
Equipos y sistema cromatográficoSe dispone de una balanza analítica (Precisa-40S-M200A, Pacisa), una centrífuga para microtubos (Heraeus), un agitador vórtex (REAX-2000, Heidolph) y un congelador (917, Forma-Scientific, Inc.).
El equipo cromatográfico (Hewlet Packard-1100) consta de desgasificador, inyector automático, bomba cuaternaria, detector UV y sistema informático (HP-Chemstation).
Calibradores y controlesPara la validación de la técnica en plasma, a partir de una disolución madre de 1.000μg/ml, se prepararon calibradores a distintas concentraciones: 5; 10; 50; 100; 150; 200; 500, y 1.000μg/ml, que posteriormente fueron diluidos con plasma a una concentración 10 veces menor. En el caso de LCR y HV, a partir de una disolución madre de 10μg/ml, se prepararon calibradores a 0,1; 0,5; 1; 3; 5; 7,5, y 10μg/ml.
Las muestras control para la determinación de LNZ en plasma se prepararon a concentraciones de 1,5; 7,5, y 75μg/ml en dicha matriz biológica. Los controles para HV y LCR se prepararon a 0,8, 4 y 8μg/ml en agua.
Preparación de la muestraEl linezolid se extrajo del plasma mediante un proceso de precipitación protéica (1:1, v/v) con ácido perclótico: a 200μl de plasma se adicionó 200μl de una disolución de HCLO4 al 3%. La mezcla se agitó durante 30 s y se centrifugó a 10.900g durante 5min. Se inyectaron 100μl del sobrenadante en el sistema cromatográfico.
Las muestras de LCR y HV no requieren de dicho proceso, por lo que se inyectó muestra directa: 100μl para LCR y 50μl de HV.
Condiciones cromatográficasLa fase móvil consistió en una mezcla de acetonitrilo y un tampón fosfato de 15mM ajustado con ácido fosfórico a pH=5 (20:80, v/v). La columna analítica fue una NOVAPAK® C18 150×3,9mm de fase reversa con precolumna (Waters, S. A.). La elución se realizó en isocrático a 1ml/min y la longitud de onda UV se fijó a λ=254nm.
Validación del métodoLa validación completa se realizó únicamente en muestras de plasma. No fue posible realizarla en las muestras de LCR y HV dada la imposibilidad de disponer de matriz biológica blanca para la preparación de calibradores y controles de calidad.
ExactitudSe evaluaron tres niveles de concentración coincidentes con las concentraciones de los controles. Se efectuaron 5 determinaciones analíticas de cada uno de los tres niveles. Se calculó según:
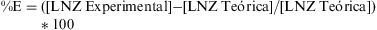
Siendo:
E: exactitud.
LNZ Teórica: concentración de LNZ teórica (μg/ml).
LNZ Experimental: concentración de LNZ obtenida (μg/ml).
PrecisiónSe evaluaron tres niveles de concentración coincidentes con las concentraciones de los controles. Se efectuaron 5 determinaciones analíticas de cada uno de los tres niveles, diariamente, durante tres días consecutivos. El estudio intraensayo se realizó con los resultados obtenidos en uno de los días anteriores. La precisión se expresó como coeficiente de variación (CV) (%) interensayo e intraensayo:
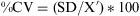
Siendo:
SD: desviación estándar de la concentración.
X′: valor medio de las determinaciones analíticas (μg/ml).
SensibilidadSe estableció mediante el límite de cuantificación. Éste se definió como la mínima concentración de LNZ, que pudo ser realmente diferenciado de la línea de base con una precisión y exactitud aceptables (<20%), tal y como se determinó en los ensayos interdía.
RecuperaciónEl cálculo de la recuperación de LNZ se realizó mediante la determinación del porcentaje de fármaco tras la extracción de muestras plasmáticas con concentraciones de LNZ conocidas frente a la inyección de la misma cantidad de fármaco en metanol/agua (1/1).
Se inyectaron paralelamente extracciones de muestras correspondientes a tres niveles de concentración, coincidentes con los controles de calidad. Se inyectaron 5 extracciones por nivel de concentración y dos muestras de patrones en metanol/agua equivalentes por concentración. Se evaluaron las áreas de los cromatogramas obtenidos:
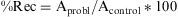
Siendo:
Rec: recuperación.
Aprobl: área del pico cromatográfico de la muestra problema.
Acontrol: área del pico cromatográfico de la muestra control.
ResultadosLas concentraciones para el estudio de linealidad fueron: 0,5–100μg/ml para plasma y 0,1–10μg/ml para líquido cefalorraquídeo y humor vítreo. La media para plasma fue de R2=0,9996. Para líquido cefalorraquídeo y humor vítreo, la media fue R2=0,9982 y R2=0,9998, respectivamente.
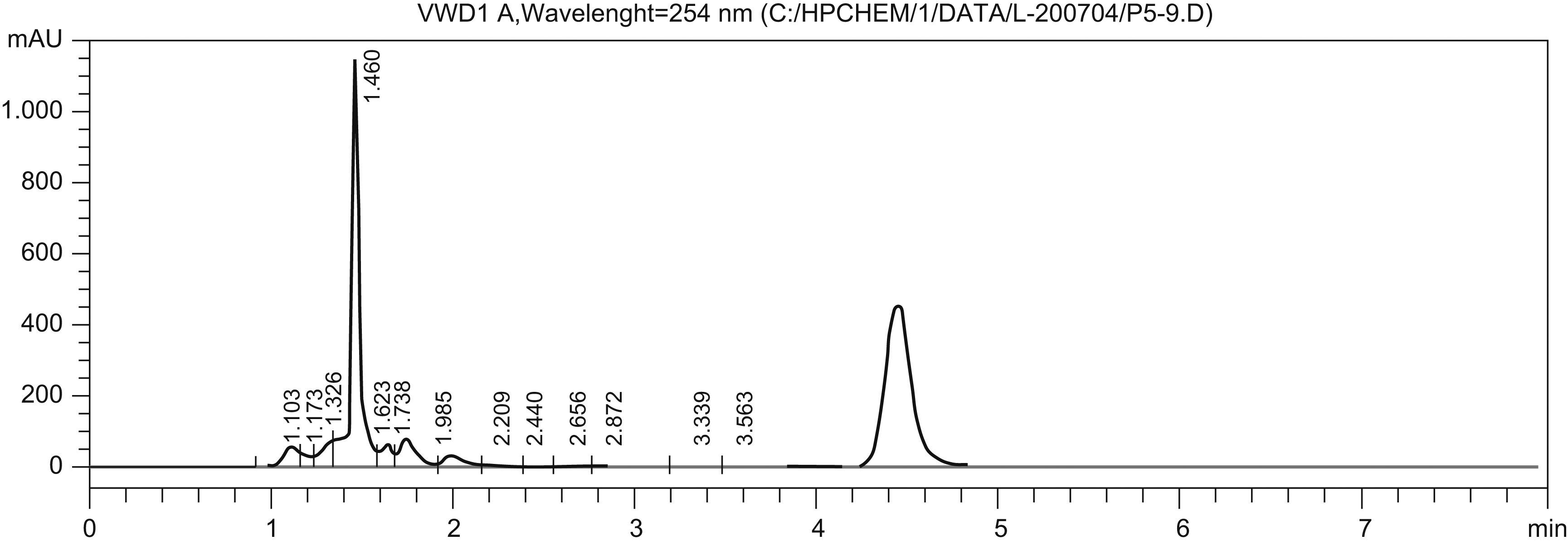
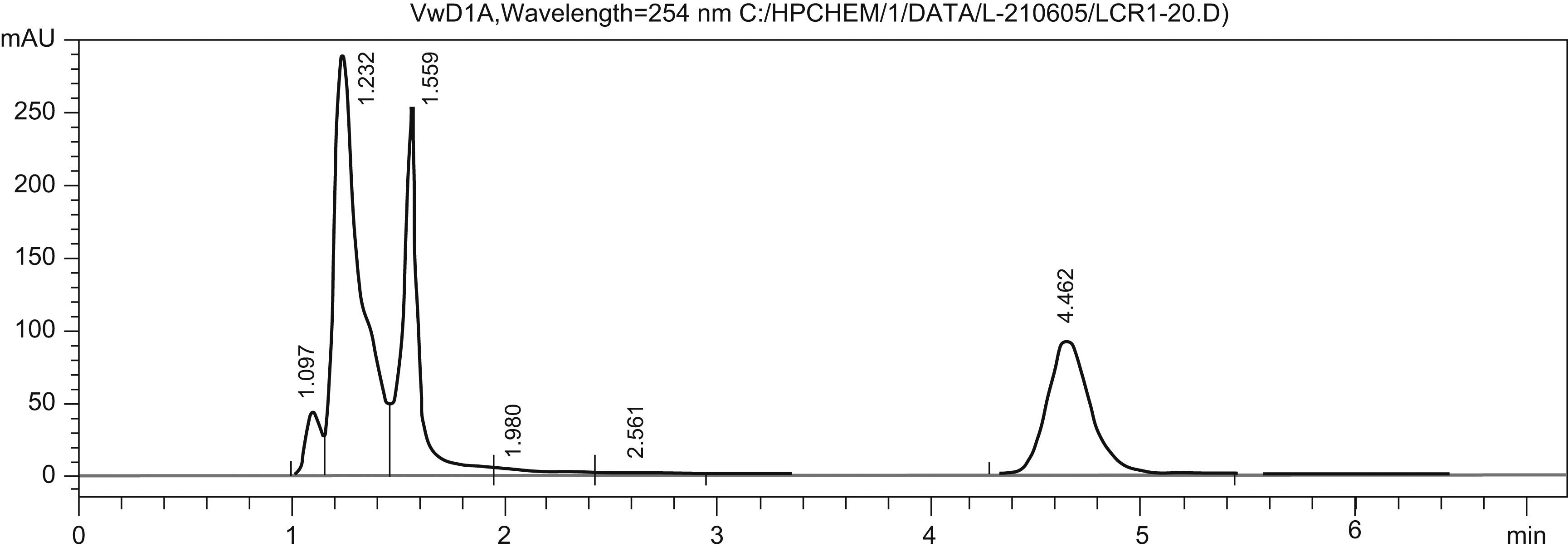
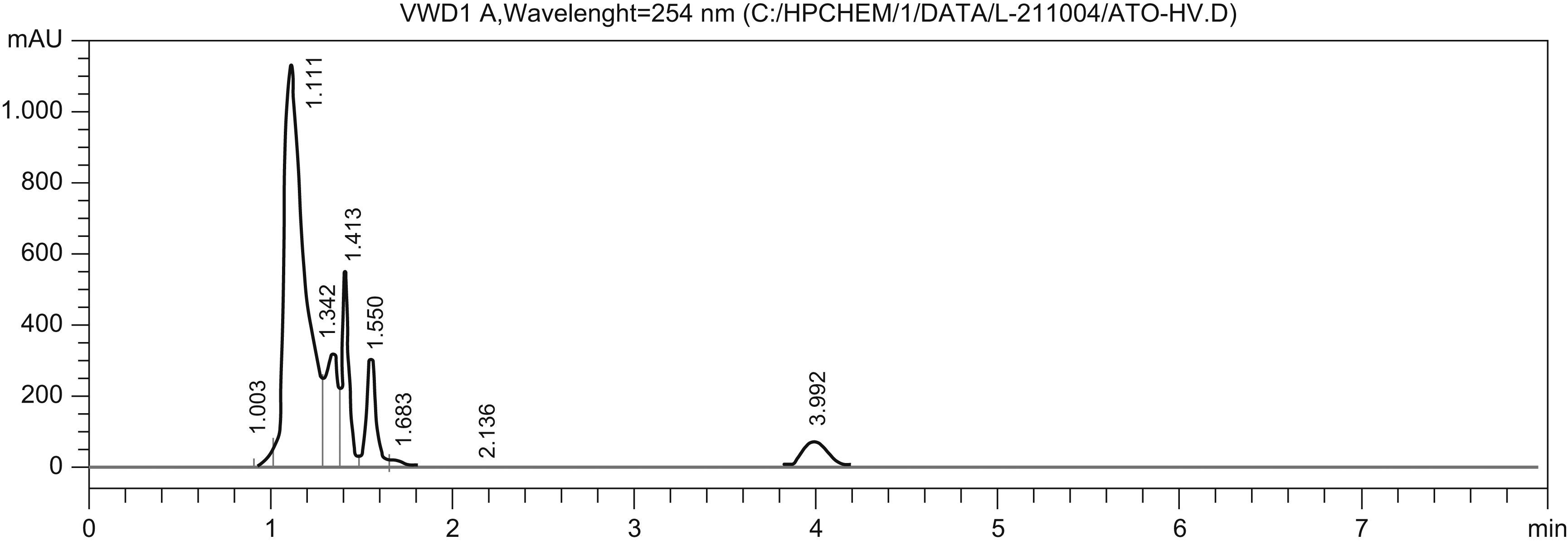
La figura 1 muestra el cromatograma que se obtuvo tras el análisis de una muestra de plasma. Las figuras 2 y 3 muestran los cromatogramas de muestras de LCR y HV, respectivamente. El tiempo de retención para el LNZ fue de 4min aproximadamente. Los resultados de exactitud para la técnica estudiada presentaron valores entre el 94,4–106,1%. Para la precisión se obtuvieron los intervalos del 0,88–6% y el 3,7–5,6% para la variabilidad intradía e interdía, respectivamente. La recuperación obtenida tras el análisis de las muestras de plasma fue del 93%.



La principal novedad de este estudio es la posibilidad de determinar y cuantificar las concentraciones de LNZ en diversas matrices biológicas, como son: plasma, HV y LCR, de manera fiable y precisa.
La diferencia principal entre el método descrito y el de Tobin et al5 es que ellos emplean una fase móvil compleja con mezcla de reactivos orgánicos para la separación cromatográfica. Por el contrario, en nuestro método la composición de la fase móvil es una simple mezcla de un tampón fosfato con un 20% de acetonitrilo. Además, en nuestro estudio empleamos una sencilla precipitación con HCLO4 para la eliminación de las proteínas de la muestra. Ehrlich et al6 y Geoffrey et al7 emplean para ello una técnica de extracción en fase sólida, lo que supone un mayor consumo de tiempo y recursos económicos. Asimismo, nuestro método no requiere de la adición de patrón interno para la cuantificación de las muestras, a diferencia de otros autores7.
De lo anteriormente descrito se deduce que las principales ventajas de nuestro método son su sencillez analítica, su rapidez, su buena recuperación analítica y su bajo coste económico.
Una limitación del estudio sería la falta de validación del método en HV y en LCR debido, principalmente, a la imposibilidad de disponer de matriz biológica blanca para preparar calibradores y controles calidad. El LCR es una solución acuosa resultante del transporte bidireccional de sustancias entre el plasma y las células del plexo coroides que se encuentran en el interior del sistema nervioso central. El LCR se produce y se reabsorbe de forma continua para mantener un volumen (150ml) y composición constante. En un individuo sano, su composición en cuanto a iones es similar a la del plasma, difiriendo de éste en tener una menor concentración de glucosa y principalmente de proteínas, por lo que puede considerarse un ultrafiltrado de plasma8. Por otra parte, el HV es una estructura gel-líquido, con un volumen aproximado de 4ml en un adulto, en la que el 99% es agua. El resto está compuesto por componentes inorgánicos, glucosa y aminoácidos en cantidades que corresponden a 1/5 de las plasmáticas9.
La ausencia de células y la baja concentración de proteínas en ambos líquidos nos permite la inyección directa al sistema cromatográfico y la extrapolación de la validación realizada en plasma sin que se produzcan problemas de interferencias analíticas. La naturaleza acuosa de estas matrices permite que se asemejen a las muestras patrón en agua analizadas durante la validación del método.
Como conclusión, podemos decir que el método se comporta de forma lineal, precisa y exacta. Además, es rápido, sensible y de bajo coste económico. La metodología descrita permite determinar concentraciones de LNZ en distintas matrices biológicas: plasma, HV y LCR, pudiendo ser utilizada como base para diversos estudios de farmacocinética clínica.
FinanciaciónFinanciado por el CIBER de Enfermedades Respiratorias 06/06/08 y el Fondo de Investigaciones Sanitarias (FIS) beca FIS PI070419.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

