




Actualmente existen seis estatinas comercializadas en España y una en proceso de comercialización. La elección de la más adecuada y de su dosis habitualmente viene determinada por el objetivo terapéutico; es decir, en el descenso de c-LDL. Se aceptan como dosis equivalentes de las distintas estatinas aquellas que consiguen el mismo porcentaje de descenso en c-LDL, sin embargo, se trata de fármacos que difieren no sólo en su potencia, sino también en sus propiedades farmacocinéticas. Una evaluación de las peculiaridades farmacocinéticas de cada estatina permite disponer de criterios para su elección, ayudando a determinar qué estatina puede ser más apropiada para un paciente en función de sus características individuales y de los medicamentos que son coadministrados.
MétodosSe revisaron las características farmacocinéticas de cada estatina y las interacciones medicamentosas que de ello se derivan.
ResultadosEl CYP3A4 es responsable del metabolismo de atorvastatina, simvastatina y lovastatina; el CYP2C9 de fluvastatina; la P-gp favorece la eliminación de atorvastina, pravastatina, simvastatina y lovastatina; y la actividad del transportador OATP1B1, que favorece el acceso al hepatocito de todas las estatinas excepto fluvastatina, es especialmente importante con rosuvastatina y pravastatina. Estas circunstancias son responsables de que aquellos fármacos que afectan a la actividad de estos isoenzimas o transportadores, bien por inhibición bien por inducción, no afecten por igual a la actividad de las diferentes estatinas.
ConclusiónA la hora de seleccionar una estatina o de intercambiar una por otra, deben tomarse en consideración las características farmacocinéticas de cada una de ellas.
The pharmaceutical industry currently offers six different statins in Spain and there is one more soon to be available. Choosing the most appropriate drug and dose is determined by the therapeutic target (reduction in LDL-C levels). Statin doses that decrease LDL-C at the same percentage are considered equivalent. Evaluating the pharmacokinetic characteristics of each statin can be useful when setting selection criteria, helping to determine which statin may be more appropriate for a patient based on their individual characteristics and on the other co-administered drugs.
MethodsWe reviewed the pharmacokinetics properties of each statin and its possible involvement in drug interactions.
ResultsCYP3A4 was responsible for the metabolism of lovastatin, simvastatin and atorvastatin; fluvastatin depends on CYP2C9; P-glycoprotein is responsible for decreased atorvastatin, pravastatin, simvastatin and lovastatin concentrations. The OATPA1B1 transporter involved in all statins’ access to the hepatocyte, except for fluvastatin, is essential for rosuvastatin and pravastatin. These circumstances cause those drugs inhibiting or inducing isoenzymes or transporters’ activity not to have the same effect on the different statins.
ConclusionThe pharmacokinetics is important when choosing the best statin and could be a limitation in the use of interchange therapeutic programmes when other drugs are present.
En los años 60, The American Heart Association comenzó a poner de manifiesto la importancia de los niveles sanguíneos de colesterol en el riesgo cardiovascular (RCV). En aquel momento los únicos fármacos disponibles para reducir el colesterol en plasma eran algunos fibratos y la colestiramina, todos ellos con una eficacia limitada1. El descubrimiento de las estatinas por Endo en el año 1973 abrió un nuevo camino en la terapia hipolipemiante, al tratarse de sustancias con elevada eficacia en la reducción plasmática de colesterol, lo que constituyó el punto de partida para una nueva línea de trabajo que vio la luz con la comercialización de lovastatina en el año 19872.
Las estatinas reducen la síntesis de colesterol en los hepatocitos mediante la inhibición de la enzima HMGCoA-reductasa. Esta reducción en la concentración intracelular de colesterol LDL (C-LDL) lleva asociada un incremento en la expresión de receptores para colesterol en la superficie de los hepatocitos, que se traduce en una mayor extracción de C-LDL de la sangre circulante, disminuyendo su concentración en la misma. Las estatinas tienen efectos adicionales sobre el perfil lipídico, ya que aumentan la concentración de colesterol HDL (C-HDL) y disminuyen la de triglicéridos (TG). A través de mecanismos de acción secundarios, disminuyen los niveles de lipoproteínas aterogénicas como la apoliproteína B100 y otras lipoproteínas ricas en TG3. Además, ejercen efectos beneficiosos a nivel cardiovascular de forma independiente de sus propiedades modificadoras del metabolismo lipídico. Estas propiedades pleiotrópicas4 pueden explicarse a través de la inhibición de la síntesis de compuestos isoprenoides no esteroideos, que también se producen a partir del ácido mevalónico, e incluyen la mejora de la función de las células endoteliales, la modificación de la respuesta inflamatoria, la reducción de la proliferación de células musculares lisas y la acumulación de colesterol.
Actualmente existen seis estatinas comercializadas en España: lovastatina, simvastatina, pravastina, atorvastatina, fluvastatina y rosuvastatina; y una en proceso de comercialización con opinión positiva de la Agencia Española de Medicamentos y Productos sanitarios (AEMP): pitavastatina. En el año 2001 cerivastatina fue retirada del mercado por la aparición de importantes reacciones adversas, principalmente rabdomiolisis.
Hoy se dispone de muy sólidas evidencias que demuestran el beneficio clínico de la disminución terapéutica de las concentraciones de colesterol total y C-LDL, tanto en la prevención primaria como secundaria de eventos cardiovasculares5-7. Actualmente, las estatinas son el tratamiento de elección para el tratamiento de la hipercolesterolemia, debido a su probada eficacia y su perfil de seguridad. Distintos ensayos clínicos han demostrado que las estatinas reducen sustancialmente la morbilidad cardiovascular y la mortalidad en pacientes con y sin cardiopatía coronaria existente3.
La eficacia terapéutica de las estatinas como hipolipemiantes se puede medir de forma objetiva a través de parámetros analíticos como TG, C-LDL, C-HDL y colesterol total (CT). La reducción del C-LDL es el objetivo principal en el tratamiento de las dislipemias, por su relación directa con el riesgo cardiovascular (RCV). En función del RCV se establecen objetivos terapéuticos, que definirán el fármaco a elegir y su posología.
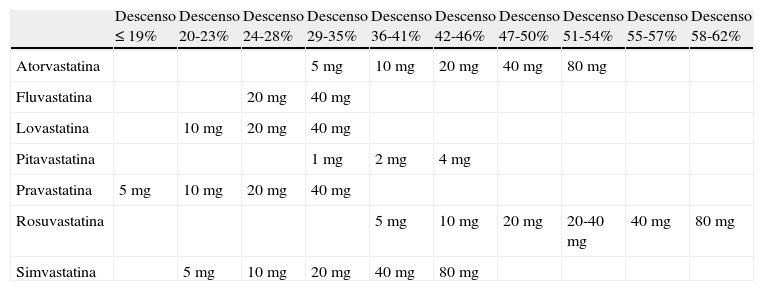
Se han publicado numerosos estudios comparativos de estatinas a nivel de seguridad y eficacia. La reducción de los eventos cardiovasculares y, en última instancia, de la mortalidad es el objetivo de la terapia con estatinas y la mejor medida de su eficacia. La mayoría de los estudios utilizan el porcentaje de reducción de C-LDL, por su relación con el RCV, como parámetro de medida más habitual a la hora de evaluar la eficacia y/o potencia de una determinada estatina8,9. Los resultados de los estudios con mayor impacto permiten elaborar una propuesta de la posología de las diferentes estatinas que puede ser necesaria para reducir el c-LDL en un determinado valor porcentual (tabla 1)8–13.
Porcentaje de descenso de c-LDL para cada dosis de estatina
| Descenso ≤ 19% | Descenso 20-23% | Descenso 24-28% | Descenso 29-35% | Descenso 36-41% | Descenso 42-46% | Descenso 47-50% | Descenso 51-54% | Descenso 55-57% | Descenso 58-62% | |
| Atorvastatina | 5 mg | 10 mg | 20 mg | 40 mg | 80 mg | |||||
| Fluvastatina | 20 mg | 40 mg | ||||||||
| Lovastatina | 10 mg | 20 mg | 40 mg | |||||||
| Pitavastatina | 1 mg | 2 mg | 4 mg | |||||||
| Pravastatina | 5 mg | 10 mg | 20 mg | 40 mg | ||||||
| Rosuvastatina | 5 mg | 10 mg | 20 mg | 20-40 mg | 40 mg | 80 mg | ||||
| Simvastatina | 5 mg | 10 mg | 20 mg | 40 mg | 80 mg |
Nota: no toda la literatura científica es unánime en el porcentaje de descenso de C-LDL que se puede obtener con cada dosis de cada estatina, por lo que esta tabla expresa una estimación de datos que no necesariamente coincide con otras tablas similares descritas en la literatura científica.
Tablas comparativas de este tipo resultan de utilidad a nivel hospitalario, donde no es habitual disponer de todos los medicamentos comercializadas, al ser una referencia a la hora de intercambiar una estatina no disponible en el hospital por una dosis equi-potente de otra incluida en la guía farmacoterapéutica (intercambio terapéutico). Los intercambios terapéuticos constituyen una práctica habitual y necesaria para la fluidez del trabajo asistencial, que, además, facilita la conciliación del tratamiento farmacológico de los pacientes durante su hospitalización14.
Condicionantes en la prescripción e intercambio de estatinasLa elección de la estatina en el momento de la prescripción, así como las sustituciones entre ellas aplicando un programa de intercambio, están sujetas a limitaciones, que tienen su origen en aspectos fundamentalmente farmacocinéticos15. El metabolismo de las estatinas está condicionado por la farmacogenética del individuo, y sus interacciones no son comunes a todas ellas, pues no todas comparten la misma vía metabólica.
Conviene resaltar el dinamismo que lleva asociado la elaboración de cualquier protocolo de intercambio terapéutico. El conocimiento científico es continuamente enriquecido con numerosos estudios, por lo que el intercambio terapéutico exige elasticidad y debe estar sujeto a una revisión continua.
MetabolismoLa capacidad de las estatinas para atravesar membranas celulares aumenta con su lipofilia. Simvastatina y lovastatina son profármacos con estructura química de lactona, que les proporciona una elevada lipofilia. Simvastatina y lovastatina presentan una baja biodisponibilidad (<5%) como consecuencia de su efecto de primer paso en la pared intestinal y a nivel hepático, debido al efecto del isoenzima 3A4 del citocromo P450 (CYP3A4) y de la glucoproteína P (P-gp). La biodisponibilidad del resto de estatinas es superior, y varía desde el 12% de atorvastatina al 51% de pitavastatina. La actividad del CYP3A4 intestinal, de las proteínas transportadoras de fármacos y el pH gastrointestinal son causas de variabilidad en la biodisponibilidad de las estatinas15. Por otra parte, las alteraciones a estos niveles provocadas por la coadministración de otros fármacos pueden contribuir a incrementar esta variabilidad.
La unión a proteínas plasmáticas es elevada en las estatinas liposolubles, siendo menor en el caso de pravastatina y rosuvastatina (las más hidrófilas). No obstante, no están descritas interacciones de relevancia clínica que tengan su origen en el desplazamiento de las estatinas de su unión a proteínas plasmáticas15.
Las formas lactona de lovastatina y simvastatina se transforman a formas ácidas (activas) por acción de esterasas. El CYP3A4 intestinal es el principal responsable de la importante eliminación presistémica de lovastatina y simvastatina, ya que inactiva tanto la forma lactona como la ácida de las mismas. El CYP3A4 hepático actúa inactivando lovastatina y simvastatina y favoreciendo la formación de metabolitos activos de atorvastatina (responsables del 70% de la actividad hipolipemiante). Fluvastatina sufre biotransformación de manera significativa por el CYP2C9 hepático. Pravastatina, rosuvastatina y pitavastatina no se metabolizan por el sistema de isoenzimas del CYP45016–19.
La P-gp da lugar a la eliminación intestinal y biliar de atorvastatina, pravastatina, rosuvastatina y las formas ácidas de lovastatina y simvastatina19. Respecto a pitavastatina, la información relativa a su afinidad por la P-gp es contradictoria; la ficha técnica aprobada por la AEMPS y algunos autores20 afirman que la pitavastatina no es sustrato de la P-gp, no obstante, algunos estudios21 señalan que la P-gp puede influir en su biodisponibilidad. Este hecho podría esclarecerse si se logra dilucidar el mecanismo de interacción entre eritromicina y pitavastatina; el antibiótico (inhibidor CYP3A4 y de la P-gp) eleva los niveles de la estatina, de tal manera que ante la ausencia de metabolismo por el CYP3A4 por parte de la misma, únicamente la inhibición de la P-gp o bien un mecanismo desconocido podrían explicar este incremento en el AUC13.
Las estatinas hidrosolubles pravastatina y rosuvastatina precisan del anión orgánico transportador de polipéptidos 1B1 (OATP1B1) para acceder al hepatocito. Lovastatina y simvastatina penetran en los hepatocitos en parte en su forma de lactona (forma liposoluble) difundiendo a través de la membrana plasmática y, en parte en su forma ácida (forma hidrosoluble) a través del OATP1B1. La actividad del OATP1B1 parece ser importante en el acceso al hepatocito de pitavastatina, presenta una relevancia aún por definir como transportador de atorvastatina, y no parece influir en el de fluvastatina13,22,23.
La actividad de las estatinas liposolubles está condicionada por el efecto de primer paso (actividad CYP3A4 y P-gp) y por la actividad del transportador OATP1B1. En el caso de las estatinas hidrófilas, su capacidad para inhibir al enzima HMGCoA reductasa depende de la actividad del OATP1B1 y de la P-gp19.
La insuficiencia hepática y la variabilidad individual (polimorfismo genético a nivel de CYP3A4, CYP2C9 y SLCO1B1 o gen codificador de OATP1B1) pueden afectar a la eficacia y seguridad de estos fármacos. La administración concomitante de otros fármacos modificadores de la actividad del CYP3A4, CYP2C9, OATP1B1 o P-gp puede generar interacciones responsables de una sobredosificación o infradosificación de las estatinas. Estos hechos exigen una precaución máxima a la hora de prescribir estos medicamentos, y suponen una limitación importante a la hora de realizar un intercambio terapéutico.
En la figura 1 y tabla 2 aparecen esquematizados y recogidos los aspectos farmacocinéticas de mayor implicación en las interacciones de las estatinas con otros fármacos.

Aspectos farmacocinéticos con mayor implicación en las interacciones potenciales de las estatinas
| Simvastatina | Lovastatina | Atorvastatina | Fluvastatina | Pravastatina | Rosuvastatina | Pitavastatina | |
| Biodisponibiliad (%) | 5 | 5 | 12 | 20-30 | 18 | 20 | 51 |
| Extracción hepática (%) | ≥ 80 | ≥ 70 | ≥ 70 | ≥ 70 | ≤ 20 | ≤ 20 | ≤ 10 |
| Metabolismo por CYP3A4 | +++ | +++ | +++ | - | + | - | - |
| Metabolismo por CYP2C9 | - | - | - | +++ | - | + | - |
| Sustrato de la glucoproteína P | SI (forma ácida) | SI (forma ácida) | SI | ? | SI | NO | NO |
| Sustrato de OATP1B1 | ++ (forma ácida) | ++ (forma ácida) | ++a | +b | +++ | +++ | +++ |
El uso de estatinas está contraindicado en pacientes con enfermedad hepática activa o con incremento injustificado y persistente de las transaminasas (aumento de tres veces el límite superior normal). En pacientes con antecedentes de enfermedad hepática y en consumidores de cantidades elevadas de alcohol deben manejarse con mucha precaución. Así, la exposición sistémica a pravastatina y sus metabolitos en pacientes con cirrosis alcohólica aumentó aproximadamente el 50%, en comparación con los pacientes con función hepática normal; lo que encierra un mayor riesgo de producir toxicidad. En todos los pacientes, está recomendado realizar pruebas de función hepática antes de iniciar el tratamiento, al incrementar sus dosis y rutinariamente con una frecuencia semestral13.
La enfermedad hepática que cursa con disfunción genera una situación compleja en el uso de fármacos en general. En el caso de estatinas la dificultad es doble, ya que no solamente es necesario prestar atención a la dosificación del propio hipolipemiante, sino que es necesario vigilar muy detenidamente el resto del tratamiento farmacológico. En esta situación, las interacciones medicamentosas pasan a tener una importancia mayor, e incluso, algunas de ellas, pueden adquirir una relevancia clínica que en condiciones normales no se produciría13.
Influencia de la genéticaLa actividad de los isoenzimas CYP3A4 y CYP2C9 presenta una gran variabilidad interindividual como consecuencia de su polimorfismo genético. A día de hoy la implicación clínica de estas variantes genéticas está todavía por demostrar24.
Los polimorfismos de SLCO1B1 (gen codificador del OATP1B1) pueden causar variabilidad en los niveles plasmáticos de las estatinas. El OATP1B1 afecta a la captación hepática de la estatina, donde va a ser metabolizada y donde ejerce su acción a nivel intracelular. Una reducida actividad del OATP1B1 puede disminuir la eficacia antihipercolesterolemiante de la estatina, e incrementar sus concentraciones plasmáticas, con el consiguiente riesgo de toxicidad muscular. Se han descrito valores de AUC de simvastatina 3,2 veces superiores en los individuos con genotipo c.521CC frente a los de genotipo c.521TT24. Debido a las propiedades farmacocinéticas de las distintas estatinas, es de esperar que los polimorfismos genéticos afecten en mayor medida a los niveles plasmáticos de pravastatina y de rosuvastatina25.
InteraccionesLa administración de estatinas de manera concomitante con otros fármacos, en ocasiones, lleva asociados riesgos de interacciones medicamentosas importantes. Estas interacciones se pueden manifestar como reducción o aumento de la actividad farmacológica o como incremento de la toxicidad, principalmente miopatía y rabdomiolisis19,26. Los pacientes con RCV, como aquellos con patología coronaria, dislipemia, diabetes, hipertensión, nefropatía, infección por VIH, trasplantados y ancianos son los que presentan mayor riesgo de sufrir interacciones clínicamente relevantes27.
Es importante mencionar que las estatinas son inhibidores muy selectivos del enzima HMGCoA reductasa, y no presentan afinidad por otros enzimas o receptores. Este hecho sugiere que, desde el punto de vista farmacodinámico, se trata de fármacos sin ningún riesgo de interacción. Las interferencias de las estatinas con otros fármacos tienen un fundamento farmacocinético, siendo los aspectos clave el metabolismo hepático (CYP3A4 o CYP2C9), la afinidad por la P-gp y por el transportador de polipéptidos OATP1B125. En la tabla 3 se recogen las interacciones farmacocinéticas más habituales de las estatinas, resaltando aquellas consideradas por los autores como clínicamente relevantes (nota: para aplicar una uniformidad de criterios, consideramos de relevancia clínica aquellas que causan un incremento del AUC [demostrado o esperado] de, al menos 2 veces, para las estatinas más potentes [atorvastatina y rosuvastatina] y de, al menos 3 veces, para el resto. También se consideraron clínicamente relevantes aquellas interacciones que implicaron una reducción de la exposición a la estatinas de, al menos, el 60%).
Interacciones más relevantes de las estatinas
Atorvastatina, fluvastatina, lovastatina y simvastatina experimentan una extracción hepática que oscila entre el 70-80%. La incidencia de miopatía en pacientes tratados con estatinas se estima en un 0,1-0,2%. Hoy en día, existen evidencias que sugieren que esta miopatía se correlaciona con el grado de inhibición del enzima HMGCoA reductasa, el cual es dosis dependiente24. La incidencia de alteraciones musculares se incrementa 10 veces en los pacientes que además de la estatina, reciben tratamiento con inhibidores del CYP3A4 como antifúngicos (ketoconazol e itraconazol), macrólidos (eritromicina y claritromicina) o antagonistas del calcio (verapamilo y diltiazem), entre otros. La mayor parte de las asociaciones de simvastatina y lovastatina con los fármacos anteriormente citados están contraindicadas; en este sentido, atorvastatina presenta un perfil de interacciones menos marcado y, a pesar de que el incremento del área bajo curva niveles plasmáticos-tiempo (AUC) sea menor, su uso concomitante con inhibidores del CYP3A4 implica una interacción con riesgo de toxicidad a nivel muscular19. Los inhibidores del CYP2C9 pueden incrementar las concentraciones plasmáticas de fluvastatina. Una interacción de relevancia clínica puede originarse cuando fluvastatina se administra de forma concomitante con fluconazol. Los potentes inductores del CYP pueden dar lugar a una disminución importante en los niveles plasmáticos de las estatinas que son metabolizadas por esta vía. Se han descrito reducciones muy marcadas en el AUC de simvastatina y lovastatina al administrarlas concomitantemente con rifampicina28 y carbamazepina29. La reducción del AUC en el tratamiento con atorvastatina es menos drástica y, en el caso de fluvastatina y pravastatina, esta interacción resulta mucho menos significativa 13,19.
Recientemente se ha descrito una interacción de relevancia clínica de simvastatina con un fármaco que afectan a la expresión génica de los isoenzimas del CYP450. Los estudios in vitro con hepatocitos humanos cultivados demostraron que la IL-6 (incrementada en la enfermedad inflamatoria autoinmune) produjo una reducción de la expresión de isoenzimas del CYP450. La administración de un fármaco con actividad antagonista de IL-6 como el tocilizumab puede provocar un incremento en el metabolismo de las estatinas13.
Interacciones a nivel de glucoproteína PLa P-gp es un transportador ATP dependiente localizado en la membrana plasmática de las células epiteliales del tracto gastrointestinal, hígado, riñón, cerebro y placenta. Actúa como una bomba excretando xenobióticos hacia el espacio extracelular, y es la responsable de los mecanismos de resistencia a ciertos fármacos como daunorubicina, vinblastina o ritonavir. El papel de las estatinas como sustratos de la P-gp está ampliamente documentado. Algunos estudios hacen referencia a una posible actividad inhibidora de la P-gp, si bien esta afirmación no cuenta con suficiente aval científico. La variación de los niveles plasmáticos que experimentan ciertos fármacos sustratos de la P-gp podría tener su origen en un mecanismo de inhibición competitiva entre diferentes sustratos del transportador. Este hecho explicaría la interacción entre digoxina y atorvastatina, que solamente se produce a dosis elevadas de la estatina (80mg/día) con un incremento del 20% en el AUC de digoxina, sustrato de gran afinidad por la P-gp30. Esta interacción podría no alcanzar relevancia clínica y, de hacerlo, se debería al estrecho margen terapéutico de la digoxina. Potencialmente pondría en situación de riesgo de toxicidad a aquellos pacientes diagnosticados de fibrilación auricular a tratamiento con el digitálico, en los que se recomienda mantener los niveles plasmáticos de digoxina cerca del límite superior del margen terapéutico. Entre todas las estatinas, atorvastatina, lovastatina, simvastatina y pravastatina (y posiblemente pitavastatina) son las que presentan afinidad por la P-gp30,31, y sus interacciones con sustratos de esta enzima como diltiazem, verapamilo, itraconazol o ciclosporina probablemente tengan un origen multifactorial con CYP3A4 y el transportador OATP1B1 como principales implicados.
Interacciones a nivel de OATP1B1El OATP1B1 es un transportador de aniones orgánicos que se encuentra en la membrana celular del hepatocito y favorece la entrada en el mismo de la mayoría de las estatinas, pero su importancia puede ser mayor con las hidrófilas (rosuvastatina y pravastatina)23. Se han descrito interacciones de diferentes estatinas con inhibidores del OATP1B1 como ciclosporina, eltrombopag y gemfibrozilo32,33. Debido a la potencia de la rosuvastatina y a la magnitud de la interacción (valores 7 veces superiores de AUC), su uso concomitante con ciclosporina está contraindicado13.
DiscusiónEl manejo de hipolipemiantes no escapa a la tendencia actual de individualizar la farmacoterapia, y la elección del fármaco adecuado debe fundamentarse en una decisión basada en un compendio de factores, entre los que se encuentran: objetivo terapéutico, patología del paciente y tratamientos concomitantes. Las estatinas son fármacos con una eficacia ampliamente demostrada, con buen perfil de seguridad y una posología cómoda (se administran en dosis única diaria). Todas estas cualidades las convierten en el tratamiento hipolipemiante más utilizado hoy en día; sin embargo, su uso no está exento de riesgos, y se debe prestar especial atención a sus potenciales interacciones cuando se administran con otros fármacos.
En el año 2001 cerivastatina fue retirada en todo el mundo por el elevado riesgo de producir rabdomiolisis. En España, hasta agosto de 2001, se habían detectado 80 casos de rabdomiolisis en pacientes tratados con cerivastatina a dosis altas, 6 de ellos mortales. Además, un 60% de los pacientes recibían tratamiento concomitante con gemfibrozilo. Esta reacción adversa hizo que inicialmente la Agencia Española del Medicamento (AEM) desaconsejase la combinación de cerivastatina y gemfibrozilo. Más tarde, debido a diversos fallecimientos que se produjeron en Estados Unidos y que se atribuyeron a la estatina, el fármaco fue retirado del mercado. A partir de ese momento surgió un debate acerca de la medida tomada por la FDA y se analizaron las posibles causas responsables de tales efectos adversos34. Parece que la dosis recomendada de la cerivastatina era, proporcionalmente, más alta que la de otras estatinas; hecho que explica su elevada eficacia a la hora de reducir las cifras de colesterol y, al mismo tiempo, la mayor incidencia de efectos adversos que, como es sabido, es dosis dependiente. Por otro lado, la administración concomitante con gemfibrozilo, puede relacionarse con una potenciación de la toxicidad muscular de ambos fármacos. Debe tenerse en cuenta que esta interacción, que hoy se conoce, se atribuye a una inhibición del transportador OATP1B1 por el fibrato, e implica a la estatina y al fibrato que llevan asociados el mayor riesgo de rabdomiolisis de sus respectivas familias.
Las interacciones farmacocinéticas de las estatinas condicionan la prescripción del hipolipemiante más adecuado y adquieren una especial relevancia en el paciente polimedicado. Precisamente los individuos con patología coronaria o con ciertas enfermedades que llevan asociado cierto RCV, a menudo manejan fármacos que interaccionan con las estatinas. Amiodarona, diltiazem y verapamilo están presentes en el tratamiento de numerosos pacientes con enfermedad cardiovascular. Por ejemplo, en un paciente tratado con diltiazem debemos plantearnos utilizar fluvastatina, pravastatina o rosuvastatina, y el nivel de colesterol deseado determinará el fármaco a elegir. Las alteraciones en el metabolismo lipídico son frecuentes en el paciente VIH, en el que los inhibidores de la proteasa son fármacos de primera línea; en esta situación, puede plantearse un tratamiento con fibratos o, en caso de precisar un fármaco más potente, recurrir a fluvastatina o pravastatina. Estas circunstancias u otras similares plantean la necesidad de introducir un tratamiento hipolipemiante en el que la elección del fármaco debe realizarse de forma minuciosa y, no necesariamente, debe concluir en una estatina.
Los avances en el conocimiento de los mecanismos farmacocinéticos de las estatinas que se han producido en los últimos años han permitido caracterizar muchas interacciones. Pravastatina se consideró durante mucho tiempo como un fármaco prácticamente de elección en aquellos pacientes que recibían tratamiento con fármacos que se comportan como inhibidores del CYP3A4, con la limitación de su potencia en la actividad hipolipemiante, inferior al resto de estatinas. Rosuvastatina se convirtió en una fármaco muy interesante debido a su ausencia de metabolismo por el CYP y su elevada potencia, la mayor de todas las estatinas. Sin embargo, se ha observado que ambos fármacos no escapan al riesgo de experimentar interacciones, ya que sufren procesos metabólicos en los que interfieren otros fármacos, como los transportadores OATP1B1 y la P-gp. Un ejemplo muy interesante que aglutina todos estos aspectos es la interacción entre ciclosporina y estatinas. El riesgo de hiperlipidemia y patología cardiovascular es elevado en el trasplantado renal, que frecuentemente recibe tratamiento con ciclosporina. Ciclosporina es un potente inhibidor del OATP1B1, P-gp y CYP3A4, e incrementa la exposición a todas las estatinas excepto fluvastatina, aunque de forma variable35. Este hecho hace necesario el ajuste de dosis en todas ellas y, en el caso de rosuvastatina, constituye una contraindicación. Todo ello deja poco margen de maniobra en el uso de estatinas, y obliga a manejar dosis bajas de las mismas, o un fármaco de menor potencia como fluvastatina, gemfibrozilo o inhibidores de la absorción de colesterol.
A nivel hospitalario las interacciones también condicionan el intercambio terapéutico. Es muy frecuente la polimedicación de los pacientes, lo cual incrementa la posibilidad de interacciones. El intercambio terapéutico de estatinas lleva asociado riesgos potenciales para el paciente, de ahí la necesidad de programas que permitan seleccionar fármacos equivalentes tanto en seguridad como en eficacia, adaptándose siempre a la situación particular de cada paciente.
La variabilidad interindividual, las potenciales interacciones, la concomitancia de ciertas patologías y de factores asociados al proceso de hospitalización, convierten tanto la prescripción como el intercambio terapéutico de estatinas en tareas altamente complejas, que deben realizarse siempre con cautela.
RecomendacionesPrescripción de estatinasLa elección de la estatina más adecuada ha de venir determinada por el nivel de C-LDL objetivo.
El uso de estatinas está contraindicado en pacientes con enfermedad hepática activa, incluyendo elevaciones inexplicables y persistentes de las transaminasas. Deben realizarse pruebas de función hepática antes de iniciar el tratamiento con estatinas, con el cambio de dosis y con el cambio de fármaco.
Durante el tratamiento se deben monitorizar signos y síntomas de toxicidad muscular.
Siempre que se prescriba una estatina o se sustituya por otra es importante analizar los demás fármacos coadministrados, debido al riesgo de interacciones con relevancia clínica.
Intercambio terapéuticoEn ausencia de alteraciones de la función hepática y de fármacos coadministrados con interacciones potenciales, todas las estatinas son intercambiables entre sí, tal y como se expresa en la tabla 1.
En presencia de fármacos inductores o inhibidores del CYP3A4, fluvastatina, pravastatina y rosuvastatina son intercambiables entre sí.
En presencia de fármacos inductores o inhibidores del CYP2C9, todas las estatinas son intercambiables entre sí, excepto fluvastatina.
En presencia de fármacos inductores o inhibidores de la activad de la P-gp, fluvastatina y rosuvastatina son intercambiables.
En presencia de fármacos inductores o inhibidores del OATP1B1, no está recomendado el intercambio terapéutico de pravastatina y rosuvastatina. Para el resto de estatinas, el intercambio debe realizarse con cautela.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

