







Introducción
La interacción entre virus y hospedador representa un proceso de extraordinaria complejidad en el que dependiendo de la virulencia del germen, sus mecanismos patogénicos y la respuesta inmunitaria se establecen infecciones agudas o crónicas en las que el equilibrio puede decantarse del lado del virus o del sujeto infectado. El virus de la inmunodeficiencia humana (VIH) origina una enfermedad crónica transmisible por vía sexual, lo que le permite propagarse y persistir en nuestra especie. Debido a su tropismo preferente por los linfocitos CD4, el VIH origina una destrucción progresiva del sistema inmunitario que tiene como expresión clínica final el sida. Además de la destrucción del sistema inmunitario, que constituye la característica más importante de la enfermedad, la infección por el VIH origina una serie de manifestaciones neurológicas y tumorales1. Por lo tanto, la fisiopatología del sida es un proceso extraordinariamente complejo en el que se encuentran implicados mecanismos patogénicos muy diferentes, algunos de los cuales no son todavía completamente comprendidos2.
Para analizar la inmunopatología del sida es necesario considerar tres aspectos esenciales:
1. Los mecanismos mediante los cuales el VIH se adapta y destruye su célula diana.
2. La respuesta del sistema inmunitario que intenta neutralizar la infección por el VIH.
3. Los mecanismos de escape que permiten al VIH eludir esta respuesta inmunitaria.
Ciclo de infección del VIH
Ciclo biológico del VIH
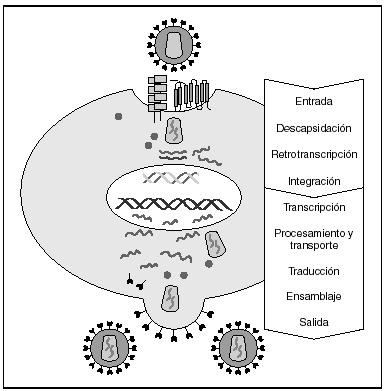
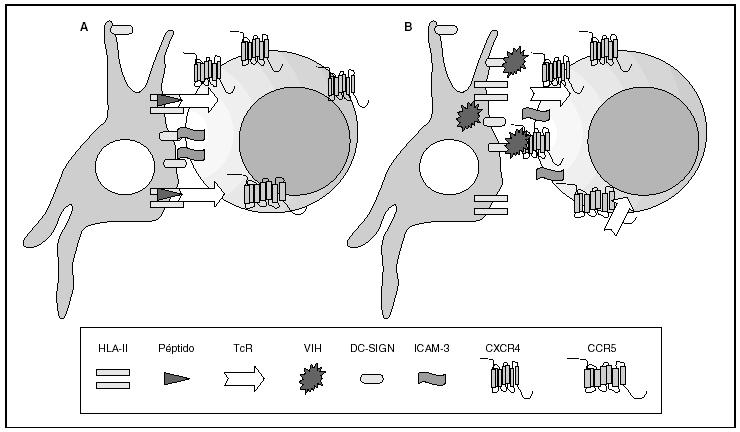
La entrada del virus en la célula representa el primer paso en su ciclo biológico3 (fig. 1) y es un proceso secuencial que se produce mediante la interacción con distintas moléculas situadas en la membrana celular. Existe en la superficie de las células dendríticas determinadas lectinas como DC-SIGN y L-SIGN a las que se adhieren con alta afinidad numerosos virus entre los que se encuentra el VIH4. Este proceso "atrapa" y concentra los viriones en la membrana plasmática (fig. 2) lo que facilita enormemente la propagación viral ya que, en este contexto, la transmisión del VIH a los linfocitos CD4 tiene una eficacia muy superior a la capacidad infectiva de partículas virales solubles no unidas a estas lectinas5.
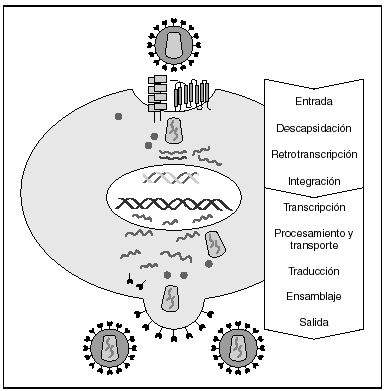
Figura 1. Ciclo biológico del VIH.
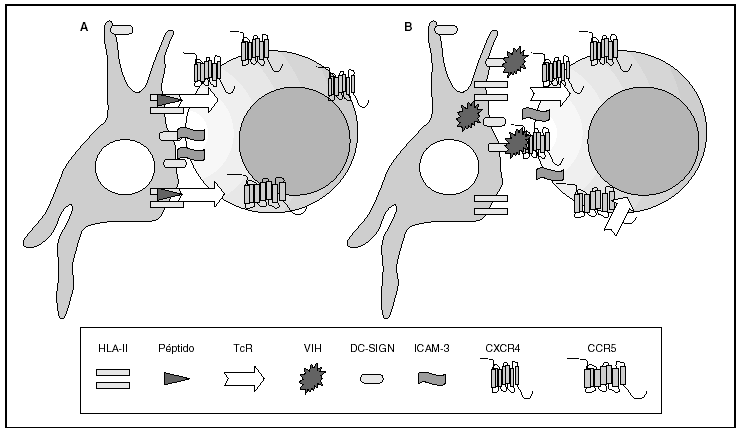
Figura 2. La sinapsis inmunitaria. Interacción entre células dendríticas y linfocitos. A) En ausencia de infección por el VIH; B) en presencia de infección por el VIH. Las células dendríticas interaccionan a través de lectinas tipo DC-SIGN con la molécula de LFA-3 presente en linfocitos y facilitando la interacción entre el receptor de la célula T (TcR) y los péptidos procesados en las moléculas HLA de clase II. Además de esta función fisiológica, en el caso de la infección por el VIH los viriones se acumulan en la superficie de las células dendríticas unidos a DC-SIGN lo que favorece la propagación del VIH en la sinapsis inmunitaria.
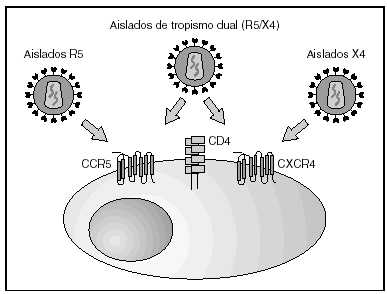
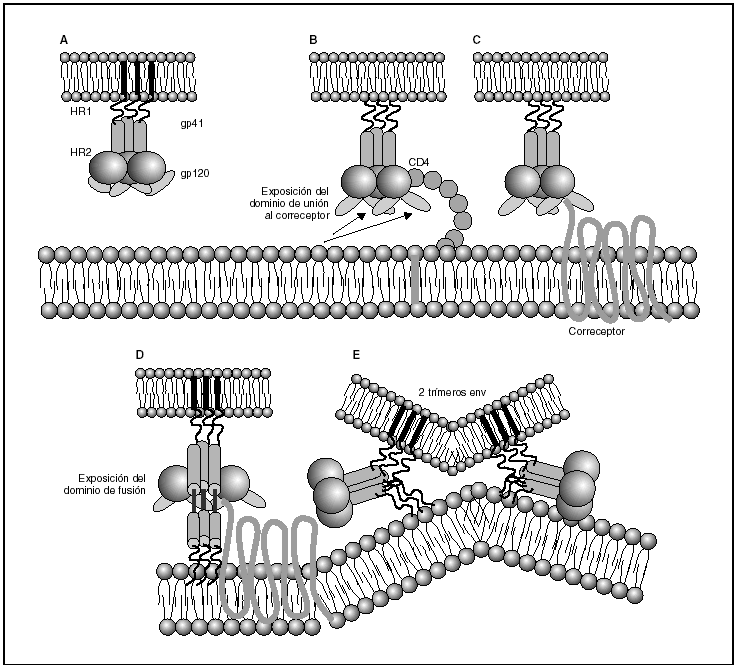
A partir de esta localización en la membrana de las células dendríticas, las partículas virales infectan a los linfocitos CD4 mediante un proceso secuencial en el que se produce la interacción con dos tipos de receptores (fig. 3): un receptor específico y común a todas las variantes del VIH, la molécula de CD46 y dos correceptores (CCR5 y CXCR4) pertenecientes a la familia de los receptores de quimiocinas7. La utilización de uno u otro correceptor depende de la secuencia de la envuelta viral que condiciona su capacidad para unirse con mayor o menor afinidad a uno o ambos correceptores. En función del correceptor utilizado, el VIH se clasifica en tres tipos de variantes: R5 (utilizan exclusivamente el correceptor CCR5); X4 (utilizan exclusivamente el correceptor CXCR4), o R5X4 (que pueden entrar en la célula utilizando cualquiera de los dos correceptores) (fig. 3). Tras la interacción con CD4, la proteína de la superficie viral gp120 experimenta un cambio conformacional que le permite la unión a los correceptores virales CCR5 y/o CXCR48 (fig. 4). Este proceso es necesario para inducir la fusión de la membrana viral con la membrana celular y lleva a la internalización de la cápside viral y su desensamblaje parcial en el citoplasma celular. Este proceso permite la retrotranscripción del genoma viral constituido por dos hebras de ARN monocatenario (fig. 4).
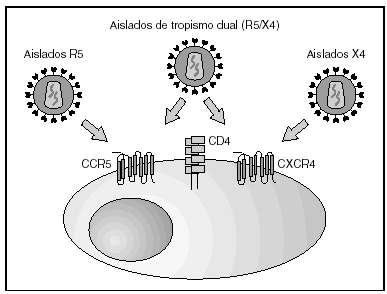
Figura 3. Receptores y correceptores del VIH.
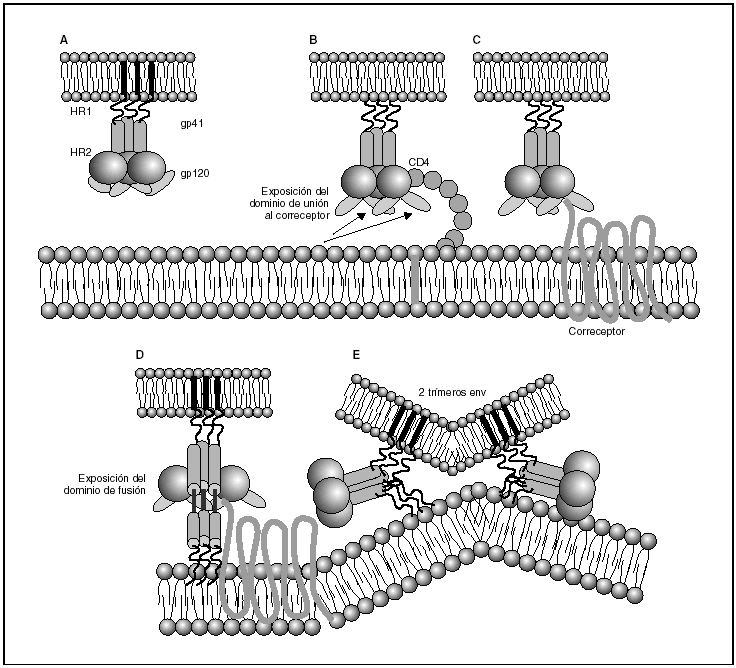
Figura 4. Dinámica de la interacción con los receptores virales y del proceso de fusión. A) Envuelta VIH; B) interacción con CD4 y cambio conformacional de la gp120; C) interacción con los correceptores; D) exposición del dominio de fusión y anclaje en las membranas viral y celular; E) formación de estructura de 6-hélices. Fusión de las membranas.
La RT o transcriptasa inversa es la enzima viral responsable de la retrotranscripción del genoma viral. Esta polimerasa convierte el ARN monocatenario en ADN bicatenario proviral. Una vez sintetizado, el ADN proviral se acopla a una serie de factores celulares y virales formando el "complejo de preintegración" que es transportado al núcleo, donde tendrá lugar la integración del ADN viral en el genoma de la célula hospedadora.
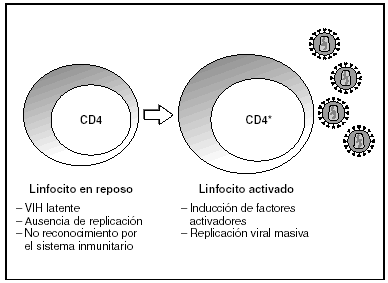
A partir del estado de integración, el VIH puede seguir un comportamiento variable: permanecer latente o experimentar una replicación masiva con el consiguiente efecto citopático sobre la célula infectada. La iniciación de la transcripción del genoma viral depende de factores celulares entre los que destaca la familia de factores de transcripción Rel/NF-kB9. Este factor no existe en forma activa en los linfocitos CD4 en estado de reposo celular y es inducido únicamente en el curso de los procesos de activación inmunológica, por lo que la replicación del VIH depende absolutamente de la activación de los linfocitos infectados10 (fig. 5). Una vez iniciada la síntesis del ARN viral, la expresión de la proteína viral Tat aumenta la tasa de transcripción del genoma del VIH y en cooperación con otros factores celulares, permite la elongación completa del ARN mensajero (ARNm) del virus11. El ARNm del VIH se sintetiza en forma de un único transcrito que debe ser transportado al citosol y procesado en ARN de distintos tamaños. Ambas etapas, procesamiento y transporte, son realizadas fundamentalmente por otra proteína viral denominada Rev. Después de su síntesis, las proteínas virales deben ser procesadas antes de ensamblarse para poder formar partículas virales maduras12. En este proceso participan proteasas celulares y la proteasa viral. Otras proteínas virales importantes en las fases finales de la infección son Vif y Vpu13. Vpu aumenta la liberación de viriones y Vif aumenta la infectividad entre 100 y 1.000 veces. La proteína Vif interacciona y neutraliza la acción antiviral de una proteína celular denominada APOBEC3G, que pertenece a la familia de enzimas de edición de ADN. En ausencia de Vif, APOBEC3G se incorpora en el virión e interfiere el proceso de retrotranscripción en la siguiente célula infectada al producir mutaciones en el ADN sintetizado por la RT, lo cual origina virus no viables y frena el proceso de propagación del VIH14. En la tabla 1 se resume la función de las distintas proteínas y del VIH.
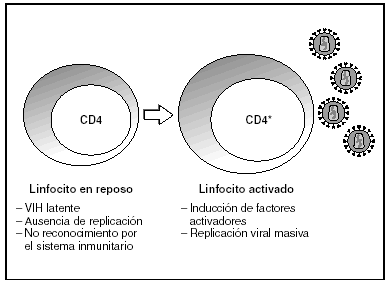
Figura 5. Activación linfocitaria y replicación del VIH.

Infección por el VIHin vivo
Etapas tempranas de la infección
En la transmisión por vía sexual las primeras células diana del virus son las células dendríticas y de Langerhans, situadas en la submucosa y los linfocitos circundantes. Debido a la presencia de DC-SIGN, los fenómenos de presentación antigénica y activación linfocitaria este microambiente es especialmente favorable a la infección de los linfocitos CD4. Una vez que se produce esta infección de la "primera estación" submucosa, la diseminación viral es inmediata y explosiva. La migración de células presentadoras y linfocitos infectados, primero a ganglios regionales y posteriormente a órganos linfoides distantes, hace que el virus se disemine a todos los órganos linfoides en una semana15. En este momento el número de linfocitos infectados es similar al observado en la fase crónica de la infección.
Fase crónica de la infección
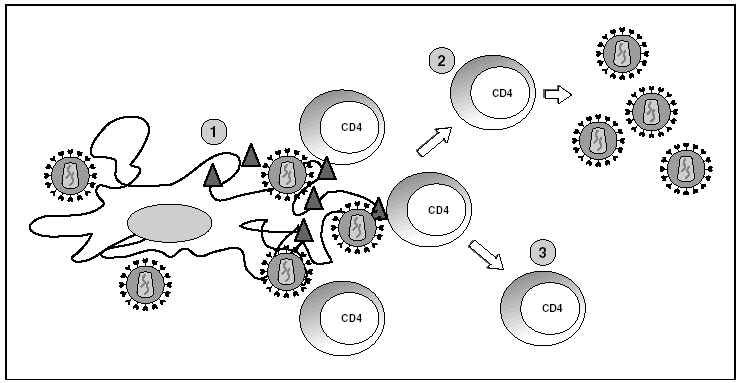
Los órganos linfoides representan el gran reservorio donde se producen los fenómenos de infección y propagación del VIH. En el estudio de los ganglios linfáticos se observa una enorme cantidad de viriones extracelulares que se disponen esencialmente en los espacios interdigitantes de las células dendríticas, en estrecho contacto con linfocitos16 (fig. 6). Esta acumulación de viriones en membrana se debe a la interacción con la lectina DC-SIGN que potencia la infectividad del VIH. Por otra parte, el VIH se detecta a nivel intracelular en los linfocitos CD4 pero sólo en una proporción muy baja de los linfocitos infectados (inferior al 1%) el VIH replica activamente mientras que la mayoría de linfocitos alberga un genoma proviral latente. La población de linfocitos CD4 activados sería la responsable de la ingente producción de viriones observada en el paciente infectado y representaría la población destruida por efecto citopático directo que tendría una semivida inferior a 24 h17. La progenie viral producida infectaría a su vez linfocitos activados, especialmente aquellos que se encuentran en fase de división para "regenerar" los linfocitos destruidos o los linfocitos CD4 específicos del VIH que al reconocer los antígenos virales se activan, lo que les hace más susceptibles a la infección viral18. La población latente representaría el reservorio de la infección viral en los linfocitos CD4 y sería inaccesible a la respuesta inmunitaria al no expresar productos virales en su membrana.
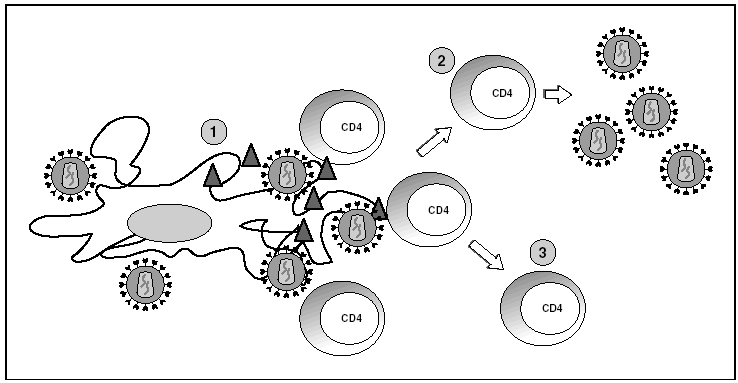
Figura 6. Infección por el VIH en órganos linfoides. 1) Virus extracelular alrededor de las células dendríticas unido a DC-SIGN; 2) linfocitos CD4 activados infectados de forma productiva; 3) linfocitos CD4 no activados infectados de forma latente.
Estadio de sida
A medida que la infección progresa, la destrucción del sistema inmunitario tiene como consecuencia un aumento en la replicación viral. Esta replicación acelerada permite una mayor generación de mutantes, con lo cual aumenta la posibilidad de evasión viral y la generación de variantes más citopáticas. La elevación de la carga viral y el rápido descenso en la cifra de linfocitos CD4 son los marcadores de una replicación "salvaje" del virus en ausencia de mecanismos de control inmunológico.
Mecanismos de linfocitopenia CD4
La destrucción de los linfocitos CD4 representa el evento más característico de la infección por el VIH19. Dada la agresiva cinética de replicación viral, se postuló inicialmente que la destrucción de los linfocitos CD4 era una consecuencia directa de la replicación viral y del consiguiente efecto citopático en la célula infectada20. Sin embargo, otros mecanismos de destrucción indirecta o de bloqueo linfocitario se hallan también implicados como causa del proceso de inmunosupresión (tabla 2), ya que la destrucción linfocitaria por efecto citopático directo no explica todos los fenómenos de disregulación inmunitaria que se observan en el sida.

Destrucción de CD4 por efecto citopático
Probablemente representa la causa más importante de destrucción linfocitaria20. Los modelos matemáticos desarrollados a partir de las modificaciones en los niveles de carga viral y linfocitos CD4 secundariamente al tratamiento con fármacos de gran eficacia antirretroviral estiman que alrededor de 108 linfocitos CD4 son destruidos diariamente por el VIH por efecto citopático directo17. Este modelo presenta críticas importantes, ya que en el mismo se presupone una distribución y tráfico homogéneo de los linfocitos entre la sangre periférica (que contiene sólo el 1% de los linfocitos totales) y los órganos linfoides. Sin embargo, algunos autores han demostrado que la acumulación de virus en los ganglios origina un fenómeno de atrapamiento de linfocitos en los órganos linfoides en torno a las células dendríticas recubiertas de viriones16. El fenómeno de aumento de CD4 que se produce en las primeras semanas tras el inicio de tratamiento antirretroviral supone de hecho una redistribución de linfocitos y no un aumento neto de éstos21. Por lo tanto, los datos actuales apuntan a que la linfocitopenia CD4 no es únicamente consecuencia de la destrucción linfocitaria por efecto citopático directo debido a la replicación del VIH y que otros mecanismos de destrucción indirecta o de bloqueo de la proliferación linfocitaria están involucrados en la profunda inmunosupresión que se produce en el curso de la infección.
Destrucción mediante mecanismos inmunitarios
Los linfocitos CD4 infectados se transforman en dianas del sistema inmunitario y al expresar péptidos virales en sus moléculas del antígeno de histocompatibilidad (HLA) de clase I son susceptibles al reconocimiento y destrucción por linfocitos citotóxicos22. En modelos experimentales se ha demostrado que la infusión de linfocitos CD8 activados frente al VIH origina una disminución en el número de linfocitos CD4 infectados23.
Destrucción secundaria a la acción de proteínas tóxicas del virus. Apoptosis
La apoptosis o muerte celular programada representa un mecanismo fisiológico mediante el cual la célula se "suicida" de forma controlada. La muerte celular inducida por apoptosis ocurre a través de dos vías: la vía extrínseca, que es activada a través de la unión en la membrana plasmática de citocinas de la familia del factor de necrosis tumoral (TNF) a receptores que activan vías bioquímicas de muerte celular. La vía intrínseca es iniciada por una serie de sensores intracelulares que modulan la actividad de proteínas que alteran la permeabilidad mitocondrial como la familia Bcl. Numerosos datos experimentales apoyan la hipótesis de que el VIH puede inducir apoptosis tanto en los linfocitos infectados como en los no infectados a través de múltiples mecanismos: activación crónica, interacción entre los receptores y la envuelta viral, efecto tóxico de proteínas virales, aumento en la expresión de ligandos citotóxicos y síntesis de citocinas por linfocitos y macrófagos (revisado en 24). De acuerdo con esta hipótesis, se ha demostrado que en los ganglios linfáticos de pacientes infectados existe in vivo una mayoría de células apoptóticas que no se encuentran infectadas25.
La proteína gp120 representa probablemente el factor proapoptótico más importante del VIH. Se ha demostrado que tanto el contacto de partículas virales como de la proteína gp120 con los receptores de los linfocitos CD4 no infectados inducen apoptosis al activar una serie de rutas metabólicas que a su vez inducen las vías extrínseca e intrínseca de la apoptosis26,27. Además de la gp120, se ha descrito que tres proteínas reguladoras, Tat, Vpr y Nef tienen un efecto proapoptótico actuando mediante distintos mecanismos. Tat activa la vía exógena y endógena de la apoptosis24 mientras que Vpr activa la apoptosis en células infectadas a través de la vía endógena28. Ambas proteínas son extremadamente tóxicas en forma soluble, en particular sobre células de sistema nervioso29, por lo que pueden ser importantes como mediadores del daño neurológico producido por la infección VIH. Por ultimo la proteína Nef desencadena apoptosis al aumentar en las células infectadas la expresión de ligandos de muerte (CD95L) que al interaccionar con moléculas de membrana en los CD8 destruyen los linfocitos citotóxicos que atacan las células infectadas30.
Bloqueo en la regeneración linfocitaria
Cuando se inicia la reconstitución inmunológica tras tratamiento antirretroviral, se observa un aumento en la cinética de división linfocitaria en la subpoblación CD4 respecto a los sujetos infectados sin tratamiento y a los pacientes seronegativos. Estos datos demuestran que durante la fase crónica de la infección el VIH bloquea la activación y proliferación de los linfocitos CD419. Únicamente con la disminución de la carga viral plasmática a niveles indetectables el sistema es capaz de entrar en ciclo de mitosis con una mayor velocidad. Este bloqueo puede producirse tanto a nivel central (timo o médula ósea) como periférico (órganos linfoides)31.
Alteraciones en la redistribución linfocitaria
La acumulación de partículas virales en los órganos linfoides, especialmente los ganglios linfáticos, origina un reclutamiento de linfocitos en estas zonas. Por tanto, la linfopenia CD4 no es únicamente debida a fenómenos de destrucción linfocitaria sino a un secuestro en los órganos linfoides19. Este fenómeno se ha descrito en otras infecciones crónicas en las que existe una gran sobrecarga y estimulación antigénica como las enfermedades por parásitos. La imagen en espejo de este fenómeno se produce tras el inicio del tratamiento antirretroviral en donde tiene lugar una rápida caída del virus extracelular unido a la membrana de las células dendríticas ganglionares que se asocia con un aumento en el número de linfocitos CD4 de memoria en sangre periférica21.
Hiperactivación y agotamiento del sistema inmunitario
Según esta hipótesis, la intensa replicación viral induciría un estado de sobrecarga antigénica y activación continua del sistema inmunológico que conllevaría un aumento en el número de ciclos de división de linfocitos, en particular de los linfocitos CD832. Esta cinética incrementada dificultaría la homeostasis normal del compartimento de linfocitos activados que serían destruidos por programas de apoptosis al cabo de un número de divisiones, disminuyendo a su vez la generación de un compartimento de linfocitos memoria.
Aunque existe una importante polémica sobre el papel de cada uno de los mecanismos previamente mencionados en la producción de linfocitopenia CD4 y es difícil cuantificar el impacto real de cada uno de estos mecanismos in vivo, probablemente todos desempeñan un papel en la linfopenia CD4 que es la característica más importante de la infección VIH.
Respuesta inmunitaria frente a la infección por el VIH
Respuesta humoral
La infección por el VIH induce una intensa respuesta de anticuerpos frente a prácticamente todas las proteínas reguladoras y estructurales del VIH33 (tabla 3). Algunos de estos anticuerpos, especialmente los dirigidos frente a gp41 y frente a los dominios variable 3 (V3) y de interacción con CD4 de la proteína gp120, tienen capacidad neutralizante in vitro34 y en experimentos de inmunoterapia adoptiva in vivo35. Sin embargo, la producción de anticuerpos con capacidad neutralizante es escasa y muy rápidamente se observa un escape viral a los mismos36. Esto probablemente se debe a que las partes expuestas y más inmunógenas de la proteína gp160 en su forma "compacta" son regiones altamente variables que inducen la síntesis de anticuerpos frente a los que el escape viral es sencillo mediante la mutación de los epítopos reconocidos. Por el contrario, los epítopos de interacción con el receptor que están mucho más conservados y que serían capaces de inducir anticuerpos neutralizantes de amplio espectro sólo se exponen cuando la proteína se despliega por la unión a CD4 (epítopos inducidos por la unión a CD4)37,38. Por otra parte, en los modelos de inmunización desarrollados hasta el momento no se obtiene de una forma consistente niveles elevados de anticuerpos neutralizantes ni su presencia se asocia de forma sistemática con protección39. Estos datos hacen que algunos investigadores planteen dudas sobre el papel de la respuesta humoral en el control de la infección por el VIH40.

Respuesta celular
En la infección por el VIH se produce una respuesta celular antiviral en distintas poblaciones: linfocitos CD4 colaboradores, linfocitos CD8 citotóxicos (CTL) y células de estirpe natural killer (NK). Sin embargo, la mayoría de los trabajos coinciden en que la respuesta CD4 y CD8 representa probablemente el mecanismo más importante de protección frente al VIH22. El estudio de la respuesta citotóxica in vitro (CTL) ha demostrado que en los pacientes seropositivos existe una expansión clonal de linfocitos CD8 con actividad citotóxica. Esta respuesta celular es particularmente intensa en pacientes en estadio de primoinfección y su intensidad correlaciona con el control de la replicación viral41. También se ha descrito una intensa respuesta CD4 y CD8 anti-VIH en algunos pacientes en el contexto de la reconstitución inmunitaria obtenida tras el tratamiento antirretroviral, especialmente en aquellos con una buena situación inmunológica antes de iniciar el tratamiento, así como en pacientes con interrupciones estructuradas de tratamiento que controlan de forma espontánea la replicación viral42. Aunque la respuesta celular CD8 es especialmente intensa frente a proteínas del core, se han descrito clones frente a distintos epítopos de la proteína de la envuelta, la transcriptasa inversa, así como frente a proteínas reguladoras. Desde el punto de vista cualitativo esta respuesta es completa en un doble sentido: se reconocen múltiples epítopos de las distintas proteínas estudiadas y el estudio del repertorio de receptores antigénicos de linfocitos T revela que se utiliza un amplio espectro de reordenamientos. Sin embargo, en pacientes infectados estables que presentan un deterioro en su evolución se ha observado un escape viral en relación con la generación de variantes virales incapaces de ser reconocidas por el repertorio linfocitario T previamente establecido43. La respuesta celular específica no se limita a los linfocitos CD8 sino que se ha demostrado que una respuesta CD4 específica es importante no sólo para la puesta en marcha de una respuesta inmunitaria eficaz frente al VIH sino por su propia actividad antiviral44. Experimentalmente los datos más concluyentes sobre el papel de la respuesta celular en el control de la replicación viral vienen de trabajos en los que la depleción de linfocitos CD8 en macacos infectados con virus de la inmunodeficiencia de simios (SIV) origina un gran aumento de la viremia y acelera la evolución a sida del animal infectado45.
Mediadores solubles
Existen numerosos factores solubles que son activos frente a la infección por el VIH como las proteínas del complemento y los interferones. Aunque probablemente estos mecanismos representen una barrera frente a la infección por el VIH son insuficientes para controlar la infección. Más importantes son probablemente otros factores como las quimiocinas, defensinas y citocinas. Desde hace años se sabe que los linfocitos CD8 de pacientes seropositivos inhiben la replicación del VIH y que esta actividad supresora corresponde a factores solubles presentes en el sobrenadante generado a partir de linfocitos CD8 activados. La descripción de que receptores frente a quimiocinas son a su vez correceptores frente a distintas cepas del VIH ha permitido caracterizar que esta actividad supresora corresponde principalmente a CC y CXC quimiocinas como RANTES, MIP-1a, MIP-1b y SDF-1/CXCL1246. Especialmente importante es el papel de SDF-1/CXCL12 en el bloqueo de la infección por las variantes X4 del VIH. Las células de los epitelios vaginal, endocervical y rectal, así como las células dendríticas producen grandes cantidades de esta quimiocina que actuaría bloqueando la propagación de variantes de tipo X447. Sin embargo, no todos los autores atribuyen esta actividad supresora a quimiocinas y postulan la existencia de otros factores supresores denominados genéricamente CAF (factores producidos por linfocitos citotóxicos activados). Algunos de estos factores han sido identificados como alfadefensinas, una familia de proteínas producidas preferentemente por células polinucleares pero también por linfocitos T cuyo mecanismo de acción no ha sido completamente caracterizado hasta este momento48.
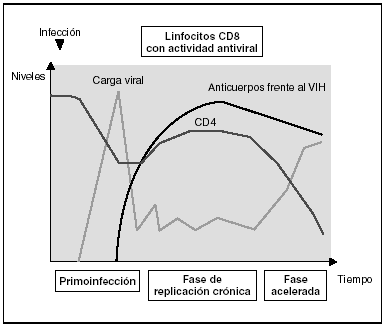
Cinética de respuesta inmunitaria en los distintos estadios de la infección (fig. 7)
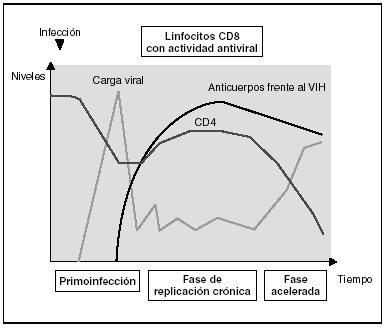
Figura 7. Evolución de los parámetros virológicos e inmunológicos en la infección por el VIH.
Primoinfección
Tras el contacto con el VIH se produce un período ventana de 4-12 semanas, que corresponde a la fase de primoinfección y durante el cual no es posible detectar la presencia de anticuerpos específicos frente al VIH a pesar de existir niveles de viremia muy elevados. Durante este período "ventana" es posible detectar actividad citotóxica frente al VIH mediante la caracterización clonal de los linfocitos CD8 del paciente. La detección de actividad antiviral celular en ausencia de anticuerpos sugiere que la respuesta celular es más precoz en el control inicial de la replicación viral que la síntesis de anticuerpos. Sin embargo, probablemente, ambos brazos de la inmunidad (humoral y celular) son importantes en el control de la replicación viral tras la primoinfección. Este control es el resultado del equilibrio entre dos factores: la virulencia de las cepas infectantes y la intensidad de la respuesta antiviral generada por el huésped. La resultante de estos dos factores se refleja en la carga viral basal del paciente tras la primoinfección, que representa un dato de enorme valor pronóstico en la evolución de la infección, ya que refleja el equilibrio alcanzado en un sujeto determinado entre el virus y su sistema inmunitario. En cualquier caso, esta respuesta antiviral es incapaz de erradicar el virus que ya se ha acantonado en las primeras horas de la infección en el organismo y se limita a contener (aunque sea en una proporción importante) la replicación viral. Se establece así una infección crónica persistente en el sujeto infectado.
Fase crónica de la infección
En la fase crónica de la enfermedad se mantienen durante años respuestas celulares y humorales intensas frente al VIH. Esta falta de atenuación de la respuesta refleja, por una parte, la intensidad y la cronicidad de la replicación viral que sigue estimulando persistentemente el sistema inmunitario y, por otra, la capacidad de éste para controlar durante largos períodos la replicación masiva que se produce a lo largo de toda la enfermedad. Sin embargo, los mecanismos de inmunosupresión y de destrucción de los linfocitos CD4 por el VIH se producen de forma persistente, y a medio plazo conllevarán una incapacidad progresiva del sistema inmunitario para contener la replicación viral. A esto se unirá la emergencia de variantes más agresivas que aumentarán la destrucción inmunológica, y desplazará el equilibrio virus-hospedador a una situación de replicación viral acelerada y de profunda inmunosupresión.
Estadio avanzado de la enfermedad
Los estadios finales de la enfermedad se caracterizan clínicamente por la aparición de infecciones oportunistas, desde el punto de vista inmunológico por la caída del número de linfocitos CD4 y, virológicamente, por la elevación de la carga viral. En esta etapa se observa un deterioro de la respuesta humoral y celular frente al VIH: disminuyen los niveles de anticuerpos frente a p24 y otras proteínas virales, decrece la tasa de anticuerpos neutralizantes, la actividad citotóxica y el número de linfocitos CD8, y se observa un deterioro en la actividad ADCC y NK. Probablemente este deterioro refleja la destrucción masiva del sistema inmunitario por una replicación viral acelerada. No hay que olvidar que el sistema inmunitario se activa de forma coordinada entre sus distintos elementos y que en la generación de la respuesta inmunitaria específica los linfocitos CD4 ocupan un lugar central. Es, por tanto, previsible que la destrucción de los CD4 origine un deterioro funcional de otras subpoblaciones celulares. Esta situación de "cataclismo inmunológico" es debida probablemente a un aumento en la cinética de replicación viral debido a la generación de mutantes de escape, incapaces de ser contenidos por el sistema. Se entra así en un círculo vicioso en el que el deterioro inmunológico progresivo permite una replicación viral más agresiva.
Mecanismos de escape viral
Variabilidad genética
La tasa de variabilidad del VIH se debe a la alta tasa de error de la transcriptasa inversa (una sustitución por 103-104 nucleótidos y ronda de copia). Esta falta de fidelidad genera una alta diversidad en las proteínas del virus que le permiten escapar al control de la respuesta inmunitaria específica. El VIH dispone por tanto de un mecanismo de escape inmunitario común a otros virus ARN en los que el alto índice de variabilidad les permite encontrar agujeros en el repertorio inmunológico.
Mutaciones en los epítopos virales reconocidos por CTL
Un aspecto no totalmente comprendido en la infección por VIH-1 es la razón por la que a pesar de respuestas inmunitarias potentes en la primoinfección no se contiene la replicación viral. Aunque se han propuestos varias explicaciones la más documentada es la del escape viral a través de mutaciones en los epítopos del virus43. El escape de la respuesta CTL se debe a mutaciones puntuales de los epítopos virales que interaccionan con el surco de las moléculas de HLA. Tanto en los pacientes primoinfectados como en modelos animales se ha demostrado cómo la mutación en un residuo conlleva un escape viral, pérdida de la respuesta CTL e incremento de la viremia. Sin embargo, en la fase crónica no se encuentra una correlación clara entre la eliminación de una determinada variante viral y la presencia de células CTL frente a ésta. Además de los datos puramente cuantitativos existen diferencias funcionales entre los CTL de pacientes progresores y no progresores como la expresión de perforinas, la producción alterada de citocinas y quimiocinas y una menor actividad del receptor del antígeno TcR por el complejo HLA/epítopo viral. Estos datos sugieren que no sólo el número de CTL sino las características cualitativas de las CTL generadas pueden ser importantes en el control de la replicación viral49.
Características bioquímicas de la envuelta viral
La estructura de la envuelta viral en su forma nativa oculta los dominios de interacción con los correceptores virales debido a la estructura trimérica y al plegamiento de la proteína (estos fenómenos son denominados exclusión oligomérica y enmascaramiento entrópico respectivamente)8. La exposición de estos epítopos conservados que son identificados por anticuerpos neutralizantes se produce en el momento de interacción entre la membrana viral y celular, un contexto en que la eficacia de los anticuerpos es menor dada su baja accesibilidad37. Un segundo mecanismo de escape más clásico es el de la mutación epitópica en las regiones hipervariables que se encuentran en el dominio externo de la envuelta viral. Sin embargo, trabajos recientes demuestran que el escape a estos anticuerpos no requiere en ocasiones la mutación epitópica, sino que puede producirse por glucosilación de los residuos y la formación de estructuras de hidratos de carbono sobre la gp120 viral denominadas "escudos glicano" y que constituyen auténticas barreras a la acción de los anticuerpos neutralizantes. A lo largo de la evolución en un paciente determinado las envueltas virales van haciéndose progresivamente resistentes a todo tipo de neutralización por anticuerpos al acumular los mecanismos de escape descritos previamente50.
Rapidez en el establecimiento de la infección
El establecimiento de la infección por el VIH después de su inoculación en el organismo por vía sexual es un proceso muy rápido. En unas horas se produce la infección de las células linfoides de la submucosa vaginal y rectal y en 7 días la infección se ha propagado a los ganglios linfáticos sistémicos en los que alcanza niveles de carga viral y proviral similares a los observados en la infección crónica15. La rapidez de instauración de estos reservorios representa un gran obstáculo para el control de la replicación viral, ya que el virus se establecerá en los linfocitos infectados en los que "persistirá" a pesar de una respuesta inmunitaria específica.
Latencia y reactivación
El VIH es capaz de infectar en forma latente sus células diana y escapa así de manera absoluta a la vigilancia inmunológica al no expresar productos virales en membrana. Por otra parte, los procesos de reactivación-reinfección son extraordinariamente rápidos10 y se producen en los centros germinales de los órganos linfoides que presentan un microambiente celular idóneo para el proceso de infección de los linfocitos activados circundantes. En confirmación de estos datos se ha demostrado que los clones linfocitarios CD4 específicos frente al VIH, que se encuentran activados, se infectan en una proporción elevada, lo que conlleva una inmunosupresión preferente de las respuestas específicas frente al VIH18. Es importante señalar que la generación continua de nuevas células latentemente infectadas a partir del compartimento de replicación viral activa genera un "archivo continuo" de los cambios del virus a lo largo de la enfermedad, incluyendo los genomas mutados de resistencia a tratamiento y las variantes de escape inmunitario. El compartimento latente no es, por tanto, estático y el VIH, en cierta manera, almacena su "historia" en el mismo, lo que constituye un mecanismo de escape tanto frente al tratamiento con antirretrovirales como frente a futuras vacunas51.
Conclusión
En los últimos años se han producido avances de gran importancia en la comprensión de la inmunopatología de la infección por el VIH. Puede decirse que la inmunología del sida se desarrolla por fin sobre bases científicas sólidas. Sin embargo, persisten dos grandes desafíos: la imposibilidad de erradicar la infección y la ausencia de vacunas preventivas o terapéuticas frente al VIH. Trasladar el conocimiento de la inmunopatogenia de la infección por el VIH a desarrollos terapéuticos que permitan contribuir a la erradicación o al menos a un mejor control de la infección y diseñar inmunógenos capaces de inducir respuestas inmunitarias frente al VIH que protejan de la infección o de la progresión de la enfermedad son los grandes desafíos de la investigación sobre el VIH en el momento actual.
Agradecimientos
Nuestro laboratorio es financiado por la Redes temáticas cooperativas de Investigación en SIDA (RIS G03/173), Fundación para la Investigación y la Prevención del SIDA en España (FIPSE 3074/99, 3120/00, 36259/01, 36453/03), Programa Nacional de Salud (SAF 2000/0028) y Comunidad de Madrid.
NOTA
Los artículos publicados en la sección "Formación Médica Continuada" forman parte de grupos temáticos específicos (antibiograma, antimicrobianos, etc.). Una vez finalizada la publicación de cada tema, se irán presentando al Sistema Español de Acreditación de la Formación Médica Continuada (SEAFORMEC) para la obtención de créditos.
Una vez concedida la acreditación, ésta se anunciará oportunamente en la Revista y se abrirá un período de inscripción gratuito para los socios de la SEIMC y suscriptores de la Revista, al cabo del cual se iniciará la evaluación, durante 1 mes, que se realizará a través de la web de Ediciones Doyma.
RELACIÓN DE SERIES ACREDITADAS:
"Actualización en antimicrobianos" (2003).
Disponible en http://www.doyma.es/eimc/formacion
5 abril / 31 oct. 2004
ANEXO 1. Avances en la inmunopatología de la infección por el VIH
1.Señale cuál de las siguientes moléculas no corresponde a un receptor del VIH:
a)DC-SIGN.
b)CD4.
c)CCR5.
d)CXCR4.
e)Todas son receptores.
2.¿Cuál de las siguientes proteínas participa en el procesamiento del ARN del VIH?
a)Tat.
b)Rev.
c)Nef.
d)Vif.
e)Vpu.
3.En relación a la proteína APOBEC3G señale cuál es la respuesta correcta:
a)Interfiere con la síntesis del ADN proviral.
b)Se asocia a la proteína Vif del VIH.
c)Un virus carente de Vif no origina una progenie infectiva en una célula que contiene APOBEC.
d)Todas las anteriores.
e)Ninguna de las anteriores.
4.En relación a DC-SIGN señale la respuesta correcta:
a)Se encuentra en la superficie de los linfocitos.
b)Tiene una afinidad selectiva por el VIH.
c)Facilita la infección de linfocitos por el VIH.
d)Todas las anteriores.
e)Ninguna de las anteriores.
5.¿Cuál de las siguientes proteínas del VIH se ha implicado en la inducción de apoptosis en linfocitos?
a)Vif.
b)Vpr.
c)Gag.
d)Pol.
e)El LTR.
6.El aumento de linfocitos CD4 que se produce en las primeras semanas de tratamiento antirretroviral:
a)Se debe a su síntesis por el timo.
b)Es secundario a la falta de destrucción linfocitaria por el VIH.
c)Son producidos en médula ósea.
d)Se produce por redistribución entre órganos linfoides y sangre periférica.
e)Se debe al efecto inmunostimulante de los fármacos.
7.Uno de los siguientes mecanismos no está implicado en el escape a la neutralización del VIH por parte de los
anticuerpos:
a)Falta de expresión de la gp120 en la superficie del virión.
b)Variabilidad en los epítopos de reconocimiento.
c)Glucosilación de los epítopos de neutralización.
d)Conformación de la proteína gp120 en la superficie del virión.
e)Formación de escudos de azúcares.
8.Señala la respuesta correcta en relación a la respuesta inmune celular frente al VIH:
a)La producen los linfocitos CD8 citotóxicos.
b)Es una respuesta protectora frente a la infección.
c)Es una respuesta que puede erradicar la infección.
d)Todas las anteriores.
e)Ninguna de las anteriores.
9.¿Qué factores solubles interfieren con la entrada del VIH en la célula?
a)Complemento.
b)Interferones.
c)Citocinas.
d)Quimiocinas.
e)Defensinas.
10.Con respecto a la latencia viral señale la respuesta correcta:
a)Es un fenómeno infrecuente in vivo.
b)Es dependiente de factores celulares.
c)Se produce en linfocitos no activados.
d)Sólo se produce en macrófagos.
e)Ninguna de las anteriores.

