







Introduction
In viral infections, the interaction between the virus and host represents an extraordinarily complex process during which, depending on the virulence of the germ, its pathogenic mechanisms and the elicited immune response, chronic or acute infections are established. In these infections, immunity can fall on the side of the virus or of the infected subject. HIV causes a sexually transmitted, chronic disease, which allows it to spread among humans. Owing to its preferential tropism for CD4 lymphocytes, HIV gradually destroys the immune system and leads to the Acquired Immunodeficiency Syndrome (AIDS). In addition to the destruction of the immune system, which is the most important characteristic of the disease, HIV infection causes a series of neurological and tumoral manifestations1. Therefore, the physiopathology of AIDS is an extraordinarily complex procedure involving very different pathogenic mechanisms, some of which are not yet completely understood2 .
In order to analyze the immune pathogenesis of AIDS, it is necessary to consider three fundamental aspects:
1. The mechanisms by which HIV adapts to and destroys its target cell.
2. The immune response, which tends to neutralize HIV infection.
3. The escape mechanisms which allow HIV to elude this immune response.
Infection cycle of HIV
Biological cycle of HIV
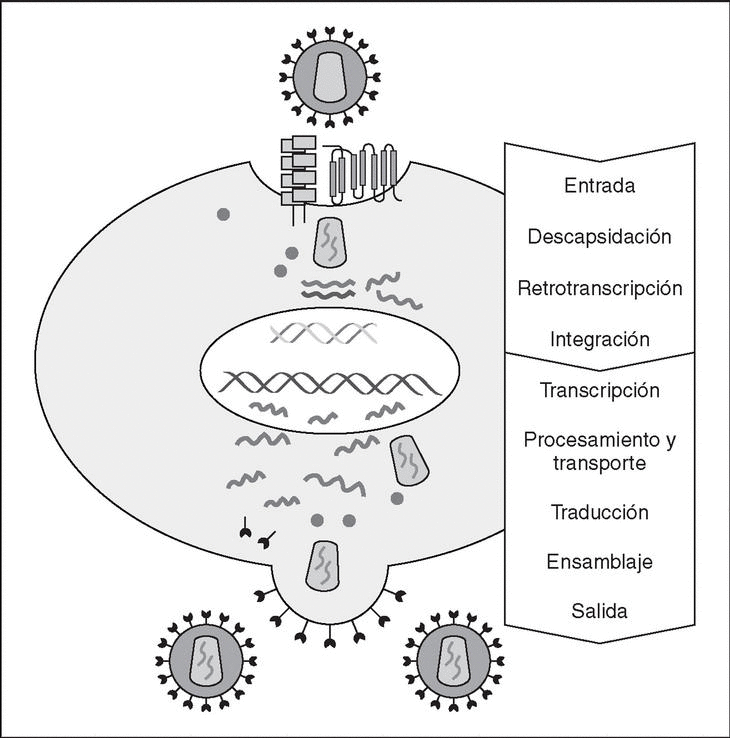
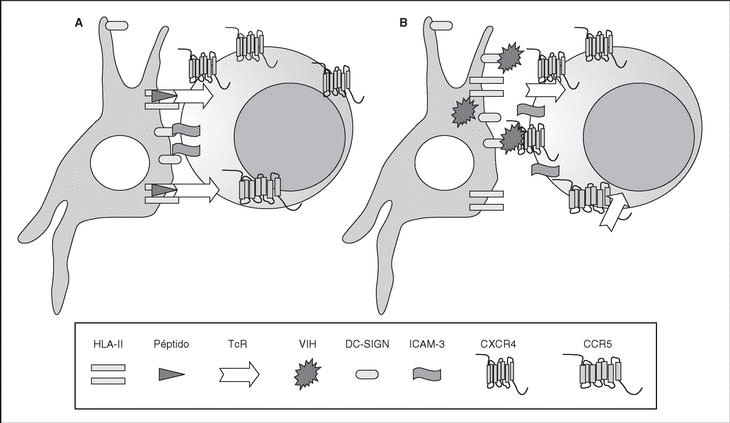
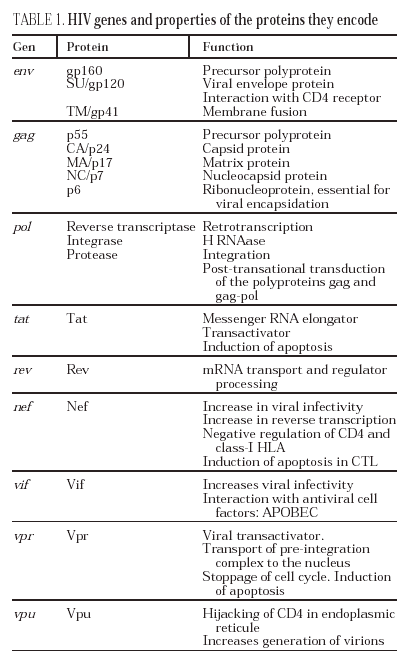
The entry of the virus into the cell is the first step in its biological cycle3 (Figure 1) and represents a sequential process which comes about through interaction with different molecules situated in the cell membrane. The surface of dendritic cells expresses C-type lectins such as DC-SIGN and L-SIGN to which several viruses, including HIV, bind with high affinity4. This process "traps" and concentrates virions in the plasma membrane, thus constituting what has been called the "immune synapsis" (Figure 2). In this cellular context, viral propagation is enhanced as transmission of HIV to CD4 lymphocytes is much more efficacious than the infective capacity of the soluble viral particles not bound to these lectins5.
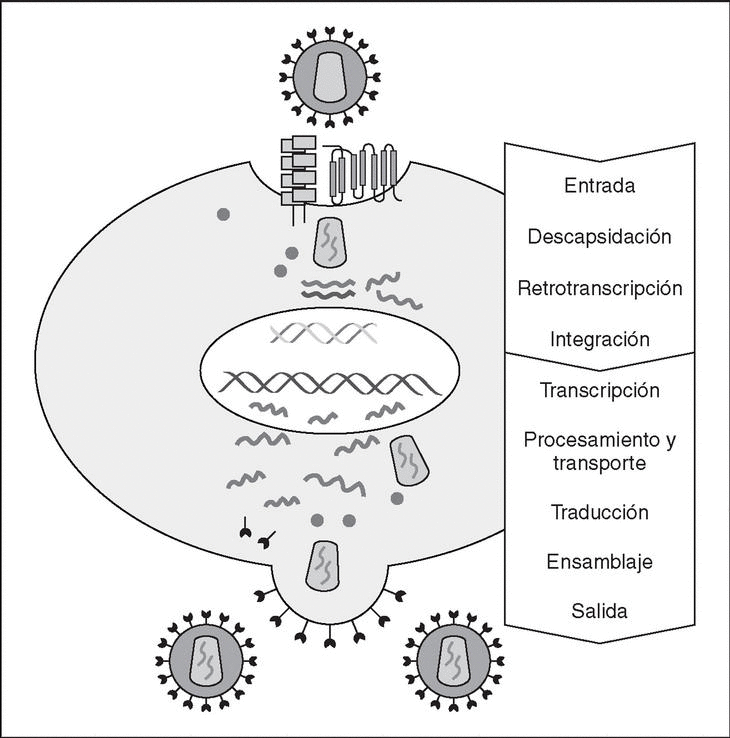
Figure 1. The HIV cycle.
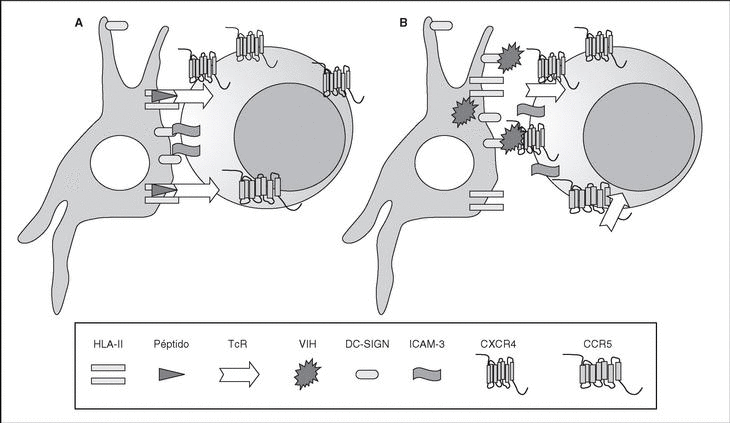
Figure 2. The immune synapsis. Interaction between dendritic cells and lymphocytes. A) In the absence of HIV infection; B) In the presence of HIV infection.Dendritic cells interact via DC-SIGN with the LFA-3 molecule present in the lymphocytes and make possible the interaction between the T cell receptor (TcR) and the peptides processed in the class II HLA molecules. In HIV infection, in addition to this physiological function, virions accumulate on the surface of the dendritic cells bound to DC-SIGN, thus favoring the spread of HIV in the immune synapsis.
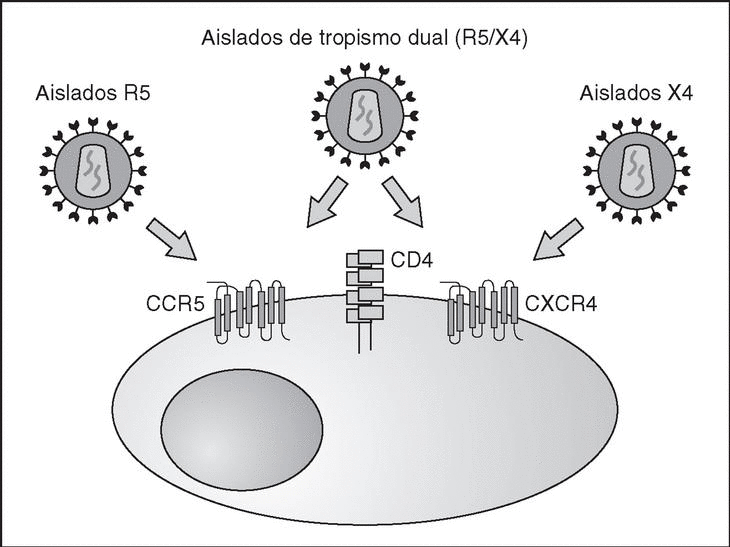
From this point, viral particles infect CD4 lymphocytes via a sequential process involving interaction with two types of receptors (Figure 3): a specific receptor which is common to all the variants of HIV variants, the CD4 molecule6, and two major co-receptors (CCR5 and CXCR4) belonging to the family of the chemokine receptors7. The use of one of the two co-receptors depends on the sequence of the viral envelope which conditions its capacity to bind with greater or lesser strength to one or both co-receptors. Depending on the co-receptor used, HIV is classified into three types of variant: R5 (which only uses co-receptor CCR5), X4 (which only uses co-receptor CXCR4) or R5X4 (which can enter the cell using either of the two co-receptors) (Figure 3). After the interaction with CD4, the viral surface protein gp120 undergoes a conformational change which allows it to bind to CCR5 and/or CXCR48 (Figure 4). This process is necessary to induce fusion of the viral and cell membranes and leads to the internalization of the viral capsid and its decapsidation in the cytosol, a process which in turn facilitates replication of the viral genome made up of the two strands of monocatenarian RNA (Figure 4).
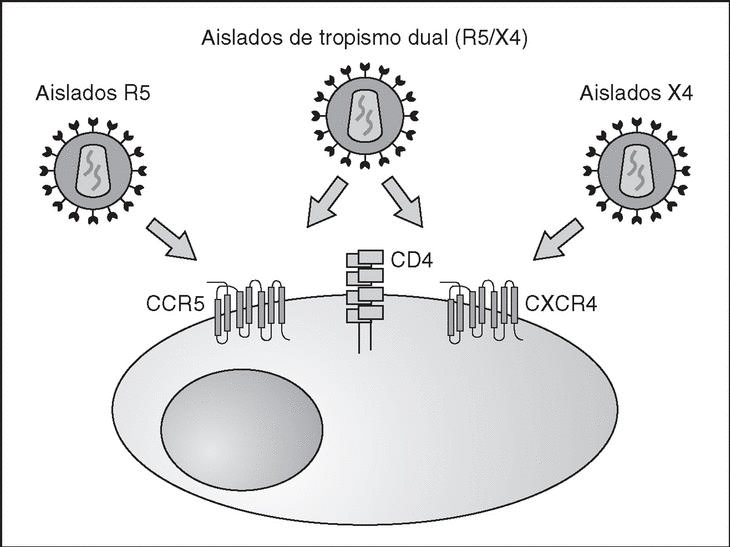
Figure 3. HIV receptors and coreceptors.
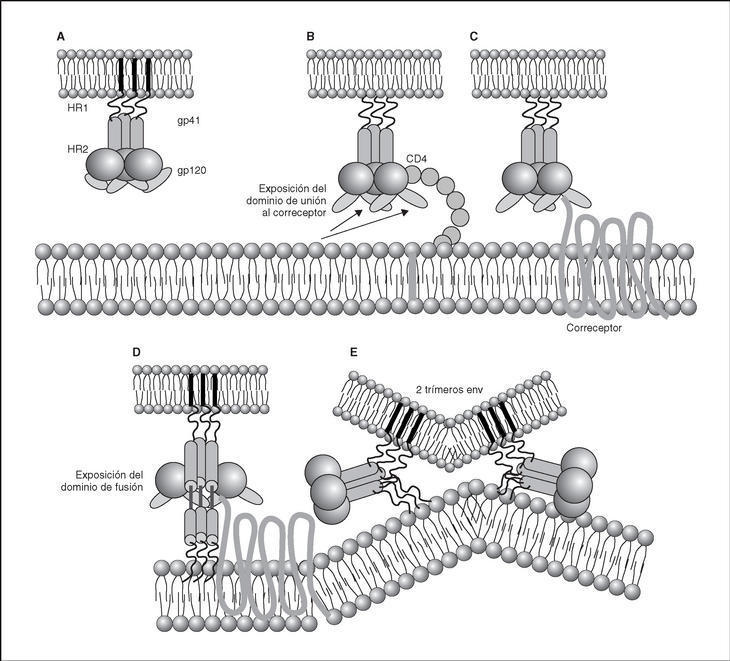
Figure 4. Dynamics of the interaction with viral receptors and the fusion process. A) HIV envelope; B) Interaction with CD4 and conformational change of gp120; C) Interaction with coreceptors; D) Release and anchorage of the fusion domain in the cell membrane; E) Formation of 6-helix structure. Membrane fusion.
Reverse transcriptase, or RT, is the viral enzyme responsible for replication of the viral genome. This polymerase converts monocatenarian RNA into proviral bicatenarian DNA, which is capable of forming part of the genome of the host cell. Once synthesized, proviral DNA binds to a series of cellular and viral factors by forming the "pre-integration complex" which is transported to the nucleus, where integration of viral DNA into the host cell genome takes place.
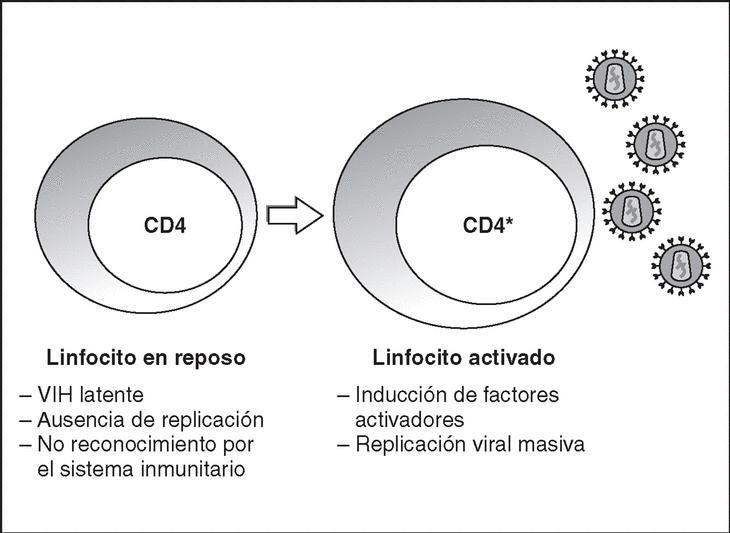
From this state of integration, the behavior of HIV can vary: it may remain latent or undergo massive replication with the subsequent cytopathic effect on the infected cell. Initiation of transcription of the viral genome depends on cellular factors, in particular the Rel/NF-*B family of transcription factors9. This factor does not exist actively in resting CD4 lymphocytes and is induced only during the processes of immunological activation. Therefore, HIV replication depends completely on the activation of infected lymphocytes10 (Figure 5). Once viral RNA synthesis has begun, expression of the viral protein Tat increases the transcription rate of the HIV genome and, in cooperation with other cellular factors, makes possible the complete elongation of viral messenger RNA (mRNA)11. HIV mRNA is synthesized as a single transcript, which must be transported to the cytosol and processed in RNAs of different sizes. Both stages, processing and transport, are essentially carried out by another viral protein known as Rev. After synthesis, the viral proteins must be processed before being assembled to form mature viral particles12. Viral and cellular proteases participate in this process. Other important viral proteins in the final phases of the infection are Vif and Vpu13. Vpu increases the release of virions and Vif increases infectivity by between 100 and 1000-fold. Vif interacts with a cell protein known as APOBEC3G, which belongs to the family of DNA-editing enzymes. In the absence of Vif, APOBEC3G is moved within the viral particle to the next target of infection where it interferes with the retrotranscription process increasing the number of mutations in the DNA synthesized by the RT. This process has as consecuence the production of unviable viruses and slows HIV propagation14. Table 1 summarizes the function of the different scion proteins of HIV.
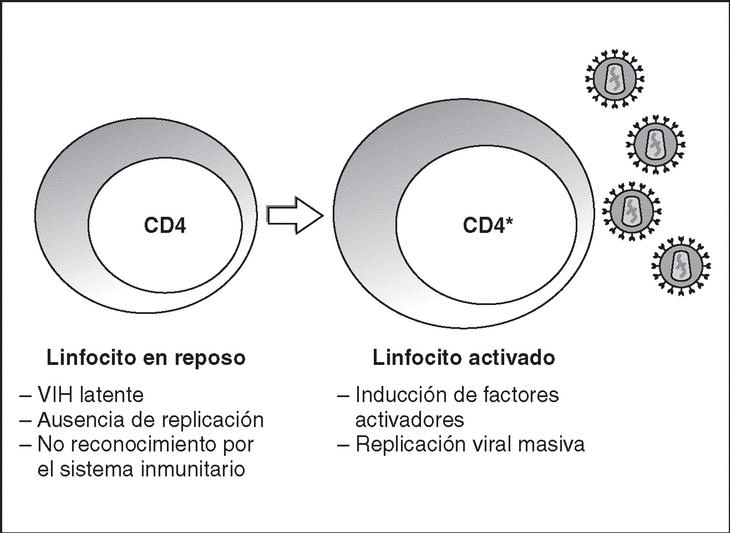
Figure 5. Lymphocyte activation and HIV replication.
HIV infection "in vivo"
Early stages
In sexual transmission, the first target cells of the virus are the dendritic and Langerhäns cells, found in the submucosa and surrounding lymphocytes. Owing to the presence of DC-SIGN, the phenomena of antigenic presentation and lymphocyte activation in this microenvironment are particularly favorable to infection of the CD4 lymphocytes. Once this "first submucosal" stage of infection begins, viral dissemination is immediate and explosive. The migration of antigen presenting cells and infected lymphocytes, first to regional ganglia and then to distant lymphoid organs, forces the virus disseminate to all the lymphoid organs in one week15. At this point, the number of infected lymphocytes is similar to that observed during the chronic phase.
Chronic phase
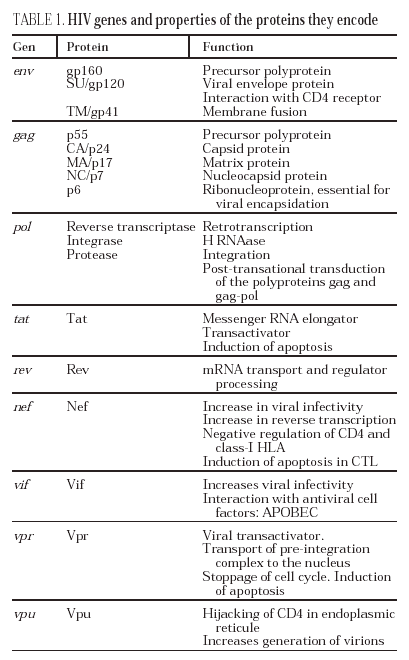
The lymphoid organs make up the huge reservoir where the infection and propagation of HIV take place. At the extracellular level of the lymphatic ganglia, we can observe a huge quantity of virions which are arranged mainly in the interdigitated processes of dendritic cells, in close contact with the lymphocytes16 (Figure 6). This accumulation of virions in the membrane is due to the interaction with the lectin DC-SIGN which boosts the infectivity of HIV. HIV can also be detected at the intracellular level in CD4 lymphocytes, but HIV only replicates actively in a very low number of infected lymphocytes (under 1%), whereas most lymphocytes harbor a latent proviral genome. This small population of activated CD4 lymphocytes is then responsible for the enormous production of virions observed in the infected patient and represents the population destroyed by the direct cytopathic effect, with a half-life below 24 hours17. The viral progeny produced then infect activated lymphocytes, especially those in the division phase, to «regenerate» the destroyed lymphocytes or the HIV-specific CD4 lymphocytes which activate when they recognize the viral antigens, thus making them susceptible to viral infection18. The latent population represents the viral infection reservoir in the CD4 lymphocytes and is essentially inaccessible to the immune response as no viral products are expressed on the cell membrane.
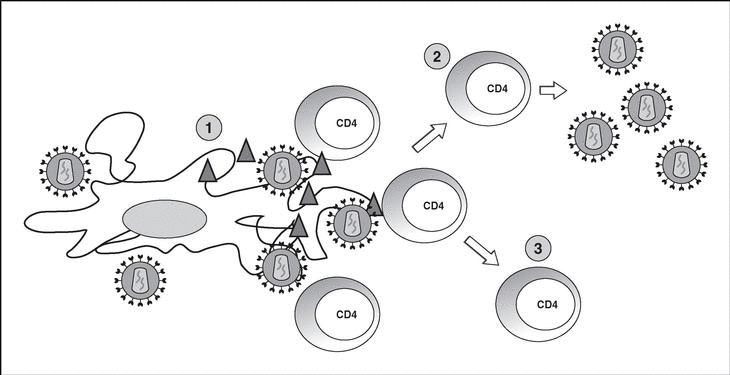
Figure 6. HIV infection in lymphoid organs. 1) Extracellular virus around dendritic cells bound to DC-SIGN; 2) Activated CD4 lymphocytes productively infected; 3) Non-activated CD4 lymphocytes latently infected.
Stage of AIDS
As infection progresses, the destruction of the immune system and lack of viral control lead to a progressive increase in HIV replication. This accelerated replication generates a larger number of mutants so that the possibility of viral escape and the generation of more cytopathic variants increases. The increase in viral load and the rapid fall in the CD4 lymphocyte count are markers of a «wild» replication of the virus in the absence of immunological control mechanisms.
Mechanisms of CD4 lymphocytopenia
The destruction of the CD4 lymphocytes is the most characteristic event of HIV infection19. Given the aggressive kinetics of viral replication, it was initially postulated that the destruction of CD4 lymphocytes was a direct consequence of viral replication and of the subsequent cytopathic effect in the infected cell20. Nevertheless, other mechanisms of indirect destruction or lymphocyte blockade are also involved as a cause of immunosuppression (Table 2), since lymphocyte destruction by direct cytopathic effect does not explain all the immune dysregulation phenomena observed in AIDS.

Destruction of CD4 by cytopathic effect
This is probably the most important cause of lymphocyte destruction20. The mathematical models developed from the modifications in viral load and CD4 lymphocyte levels after highly active antiretroviral therapy calculate that HIV destroys around 108 CD4 per day by direct cytopathic effect17. This model is open to criticism, since, it presupposes a homogeneous traffic and distribution of lymphocytes between peripheral blood (which contains only 1% of total lymphocytes) and the lymphoid organs. Nevertheless, some authors have shown that the accumulation of viruses in the ganglia produces a phenomenon of lymphocyte trapping in the lymphoid organs around the virion-covered dendritic cells16. The increase in CD4 during the first weeks after the beginning of antiretroviral therapy is, in fact, a redistribution of lymphocytes and not a net increase in the number of CD4 cells21. Therefore, current data point to CD4 lymphocytopenia as not being the only consequence of lymphocyte destruction by direct cytopathic effect due to HIV replication. Other mechanisms of direct destruction or blockade of lymphocyte proliferation also seem to be involved in the deep immunosuppression which takes place during the course of the infection.
Destruction by immune mechanisms
Infected CD4 lymphocytes become targets of the immune system and, by expressing viral peptides in their class-I HLA molecules, are susceptible to recognition and destruction by cytotoxic lymphocytes22. Experimental models have shown that the infusion of activated CD8 lymphocytes against HIV leads to a reduction in the number of infected CD423.
Destruction following the action of toxic viral proteins. Apoptosis
Apoptosis, or programmed cell death, is a physiological mechanism by which the cell «commits suicide» in a controlled way. Apoptosis-induced cell death occurs via two pathways: the extrinsic pathway, which is activated in the plasma membrane by binding of cytokines from the TNF family to receptors which in turn activate the biochemical pathways of cell death. The intrinsic pathway is activated by series of intracellular sensors, such as the Bcl-2 family, which modulate the activity of proteins which in turn alter mitochondrial permeability. There is ample experimental data to support the hypothesis that HIV can induce apoptosis both in infected lymphocytes and in non-infected lymphocytes via several mechanisms: chronic activation, interaction between receptors and the viral envelope, toxic effect of viral proteins, increase in the expression of cytotoxic ligands and synthesis of cytokines by lymphocytes and macrophages (reviewed in 24). According to this hypothesis, it has been shown that, in the lymphatic ganglia of infected patients, there exist in vivo a majority of apoptotic cells which are not infected25.
Protein gp120 is probably the most important pro-apoptotic factor in HIV. It has been demonstrated that both the contact of viral particles and that of recombinant gp120 with the receptors of non-infected CD4 lymphocytes induce apoptosis by activating a series of metabolic routes which in turn induce the extrinsic and intrinsic pathways of apoptosis26,27. In addition to gp120, it has been reported that three regulatory proteins, Tat, Vpr and Nef, have a pro-apoptotic effect by acting through different mechanisms. Tat activates the exogenous and endogenous pathway of apoptosis24 whereas Vpr activates apoptosis in infected cells via the endogenous pathway28. Both proteins are extremely toxic in soluble form, especially for cells of the central nervous system29. Therefore, they may play an important role as mediators of the neurological damage caused by HIV infection. Lastly, the protein Nef increases the expression of death ligands (CD95L) in infected cells, a process that triggers apoptosis in surrounding viral-specific CTLs through the cross-linking of Fas/CD95 in these cells30.
Blockade in lymphocyte regeneration
When immune reconstitution begins after antiretroviral therapy, there is an increase in the kinetics of lymphocyte division in the CD4 subpopulation in naive subjects and seronegative patients. These data show that, during the chronic phase of the infection, the entry of CD4 lymphocytes during the division cycle is blocked19. The system is only able to enter the cycle of mitosis with greater speed thanks to a reduction in viral load in plasma to undetectable levels. These data point to the existence of a blockade in lymphocyte activation and proliferation due to HIV infection. This blockade can occur centrally (thymus or bone marrow) or in the periphery (lymphoid organs)31.
Alterations in lymphocyte redistribution
The accumulation of viral particles in lymphoid organs, especially in lymph nodes, leads to recruitment of lymphocytes in these areas. Therefore, CD4 lymphopenia is not only due to lymphocyte destruction phenomena, but to a «hijacking» in the lymphoid organs19. This phenomenon has been reported in other chronic infections in which there is notable antigenic stimulation and overload, as is the case in parasitic diseases. The mirror image of this phenomenon appears after the start of antiretroviral therapy, during which there is a rapid fall in the number of extracellular viruses bound to the membrane of dendritic cells in lymph nodes, leading to redistribution and a sharp increase in the number of CD4 memory lymphocytes in peripheral blood21.
Hyperactivation and exhaustion of the immune syste
According to this hypothesis, intense viral replication induces a state of antigenic overload and continuous activation of the immune system which in turn leads to an increase in the number of lymphocyte division cycles, especially of CD8 lymphocytes32. The increased kinetics hinders normal homeostasis of the activated lymphocyte compartment. These lymphocytes are destroyed by apoptosis after a certain number of divisions, at the same reducing the generation of a memory lymphocyte compartment.
Although there is an important controversy surrounding the role of each of the previously mentioned mechanisms in the production of CD4 lymphocytopenia, and it is difficult to quantify the real impact of these mechanisms "in vivo", they all probably play a role in CD4 lymphopenia, which is the most important characteristic of HIV infection.
Immune response to HIV infection
Humoral response
HIV infection induces an intense antibody response to practically all the regulatory and structural proteins of HIV33 (table 3). Some of these antibodies, especially those which target the variable regions of the gp120 and those domains involved in the interaction with CD4 and co-receptors, have neutralizing capacity in vitro34 and in adoptive immunity in vivo experiments35. Nevertheless, the production of antibodies with neutralizing capacity is scarce and viral escape from these is rapidly observed36. This is probably due to the fact that the exposed and most immunogenic parts of gp160 in its "compact" form are highly variable regions which induce synthesis of antibodies whose neutralizing effect can be easily overcome by mutation in the recognized epitopes, thus leading to viral escape from antibody neutralization. On the contrary, receptor interaction epitopes which are much better conserved and which would be capable of inducing broad-spectrum neutralizing antibodies would only be exposed when the protein is displayed by binding to CD4 (epitopes induced by binding to CD4)37,38. Furthermore, in the immunization models developed to date, high levels of neutralizing antibodies have not been consistently obtained, and their presence has not been associated systematically with protection39. These data have raised doubts among some researchers about the role of the humoral response in the control of HIV infection40.

Cellular response
In HIV infection, there is an antiviral cellular response in different populations: helper CD4 lymphocytes, cytotoxic CD8 lymphocytes (CTL) and NK stock cells. Most studies agree that the CD4 and CD8 responses probably represent the most important protective mechanism against HIV progression22. The study of the cytotoxic response in vitro (CTL) has shown that, in seropositive patients, there is a clonal expansion of CD8 lymphocytes with cytotoxic activity. This cellular response is particularly intense in patients with primary infection and its intensity correlates with the control of viral replication41. An intense anti-HIV CD4 and CD8 response has also been reported in some patients in the setting of immune constitution after antiretroviral therapy, especially in those with a favorable immunological situation before beginning treatment, as well as in patients with structured treatment interruptions that spontaneously control viral replication42. Despite the fact that the CD8 cellular response is particularly intense against core proteins, there have been reports of clones against different epitopes of the envelope protein, reverse transcriptase and regulatory proteins. From a qualitative point of view, this response is complete in two senses: multiple epitopes of the different proteins studied are recognized and the study of the repertoire of T lymphocyte antigenic receptors reveals that a broad spectrum of TcR rearrangements is used. Nevertheless, in stable infected patients who present a deterioration in their evolution, viral escape has been observed with the generation of viral variants incapable of being recognized by the previously established T lymphocyte repertoire43. The specific cell response is not limited to CD8 lymphocytes - it has been shown that a specific CD4 response is important not only for triggering an efficacious immune response to HIV but due to its own antiviral activity44. The most conclusive data on the role of the cell response in the control of viral replication come from studies on CD8 lymphocyte depletion in SIV-infected macaques, which leads to a huge increase in viremia and accelerates the evolution to AIDS in the infected animal45.
Soluble mediators
Numerous soluble factors, such as complement proteins and interferons, are active against HIV. Even if these mechanisms probably represent a barrier against HIV infection, they are insufficient. Other factors, such as chemokines, defensins and cytokines are probably more important. For many years it has been known that the CD8 lymphocytes of seropositive patients inhibit HIV replication and that this suppressor activity corresponds to soluble factors present in the supernatant generated from activated CD8 lymphocytes. The report that chemokine receptors are in turn co-receptors against different strains of HIV, has made it possible to characterize this suppressor activity as corresponding mainly to CC and CXC chemokines such as RANTES, MIP-1α, MIP-1β and SDF-1/CXCL1246. Particularly important in the blockade of infection by the X4 variants of HIV is the role of SDF-1/CXCL12. Cells of the vaginal, endocervical and rectal epithelia, as well as dendritic cells, produce large quantities of this chemokine, which may act by blocking the propagation of X4-type variants47. Nevertheless, not all authors attribute this suppressor activity to chemokines, but rather postulate the existence of other suppressor factors known generically as CAF (factors produced by activated lymphocytes). These factors have been identified as α-defensins, a family of proteins produced more by polynuclear cells but also by T lymphocytes. The mechanism of action of defensins has not been completely characterized until now48.
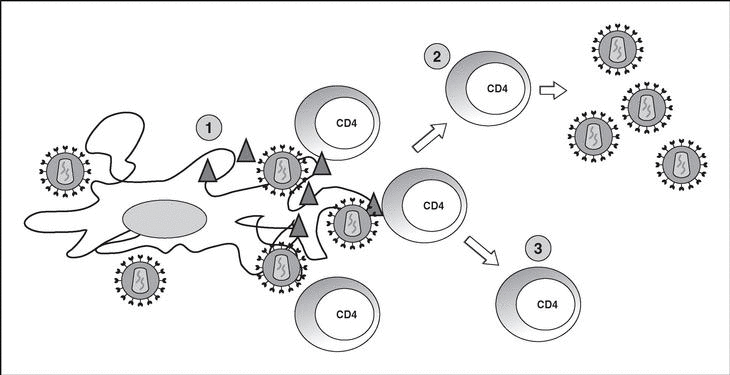
Immune response kinetics at the different stages of infection (Figure 7)
Figure 7. Evolution of virological and immunological parameters in HIV infection.
Primary infection
After contact with HIV, there is a window period of 4-12 weeks, which corresponds to the primary infection phase and during which it is not possible to detect the presence of HIV-specific antibodies despite very high viremia levels. During this window period it is possible to detect cytotoxic activity against HIV by clonal characterization of the patient's CD8 lymphocytes. The detection of cellular antiviral activity in the absence of antibodies suggests that the cell response is earlier and more important in the initial control of viral replication than the synthesis of antibodies. Both types of immunity (humoral and cellular) are probably important in the control of viral replication after primary infection. This control is the result of the balance between two factors: the virulence of the infecting cells and the intensity of the antiviral response generated by the host. The result of these two factors is reflected in the baseline viral load of the patient after primary infection, which provides extremely valuable prognostic information about the outcome of the infection, since it indicates the balance reached in a specific subject between the virus and the immune system. In any case, this antiviral response is incapable of eradicating the virus which has already lodged itself in the body during the first hours of infection and is limited to containing (even if it is an important proportion) viral replication. A persistent chronic infection is thus established in the infected subject.
Chronic phase
During the chronic phase of the disease, intense cellular and humoral responses are maintained for years against HIV. This lack of attenuation in the response reflects, on the one hand, the intensity and chronicity of viral replication which continues to stimulate the immune system, and, on the other, the ability of the immune system to control for long periods the massive replication which occurs throughout the disease. However, the mechanisms of immunosuppression and destruction of CD4 lymphocytes by HIV are persistent and, in the medium term, they imply a progressive inability of the immune system to ion and displace the virus-host balance towards a situation of accelerated viral replication and profound immunosuppression.
Advanced stage
The final stages of the disease are characterized clinically by the appearance of opportunistic infections, in immunological terms, by the fall in the number of CD4 lymphocytes and, in virological terms, by the increase in viral load. During this stage, a deterioration in the humoral and cellular response to HIV is observed: levels of p24 antibodies and other viral proteins fall, the rate of neutralizing antibodies, cytotoxic activity and the number of CD8 lymphocytes decreases, and a deterioration of ADCC and NK activity can be observed. This deterioration probably reflects the mass destruction of the immune system by accelerated viral reproduction. We must not forget that the immune system is activated in a coordinated way between its different elements, and that they play a central role in the generation of the CD4 lymphocyte-specific immune response. Therefore, the destruction of CD4 can be expected to lead to a functional deterioration of other cellular subpopulations. This «immunological cataclysm» is probably due to an increase in the kinetics of viral replication stemming from the generation of escape mutants, which the system cannot contain. A vicious circle is then created in which progressive immunological deterioration leads to more aggressive viral replication.
Viral escape mechanisms
Genetic variability
The variability rate of HIV is due to the high error rate of reverse transcriptase (one substitution per 103-104 nucleotides and round of copy. This lack of fidelity generates a high diversity in viral proteins, which makes it possible to escape the control of the specific immune response. HIV therefore has an immune escape mechanism, common to all RNA viruses in which the high variability index allows them to find holes in the immunological repertoire.
Viral epitope mutations recognised by CTLs
One aspect of HIV-1 infection which is not totally understood is the reason why viral replication is not contained, despite potent immune responses during primary infection. Several proposals have been put forward, although the most widely reported is that of viral escape via mutations in the viral epitopes43. CTL escape response is often due to ad hoc mutations of viral epitopes which interact with the groove of the HLA molecules. Both in patients with primary infection and in animal models, it has been shown how the mutation in a residue involves a viral escape, loss of the CTL response and increase in viremia. However, during the chronic phase, there is no clear correlation between the elimination or presence of a specific viral determinant and the presence of CTL cells against it. In addition to purely quantitative data, there are functional differences between the CTL of progressors and non-progressors such as the expression of perforins, the altered production of cytokines and chemokines and a lower activity of TcR antigen receptor by the HLA viral epitope complex. These data suggest that not only the number, but also the qualitative characteristics of the CTL generated may be important in the control of viral replication49.
Biochemical characteristics of the viral envelope.
The structure of the viral envelope in its native form hides the domains of interaction with viral co-receptors owing to the trimeric structure and folding of the protein (oligomeric exclusion and entropic masking)8. The exposure of these conserved epitopes which are identified by neutralizing antibodies occurs at the time of interaction between the viral and cellular membrane, a setting in which the efficacy of the antibodies is lower, given their low accessibility37. A second, more classic escape mechanism is epitopic mutation in the hypervariable regions situated in the external domain of the viral envelope. However, recent studies show that escape from these antibodies sometimes does not require epitopic mutation, rather it can come about by glycosylation of the residues and the formation of carbohydrate structures on viral gp120 known as "glycan shields", which represent authentic barriers to the action of the neutralizing antibodies. Throughout evolution in a specific patient, the viral envelopes gradually become resistant to all types of neutralization by antibodies by accumulating the previously described escape mechanisms50.
Speed in the establishment of infection
Establishment of infection by HIV after inoculation in the body through sexual transmission is a very rapid process. In a few hours, the lymphoid cells of the rectal and vaginal submucosa become infected and in seven days infection spreads to systemic lymphatic ganglia where it reaches a proviral and viral load similar to that of chronic infection15. The speed with which these reservoirs appear is an important obstacle to the control of viral replication, since the virus will be established in the infected lymphocytes where it will "persist" despite a specific immune response.
Latency and reactivation
HIV is capable of infecting its target cells latently and thus completely escapes immunological surveillance by not expressing viral products in the membrane. Furthermore, reactivation-reinfection processes are extraordinarily fast10 and they take place in the T-cell parafollicular areas of lymphoid organs, which present an ideal cellular microenvironment for the process of infection and viral propagation. As a confirmation of these data, it has been shown that HIV-specific CD4 lymphocyte clones are infected at a higher rate, implying a preferential immunosuppression of the HIV-specific responses18. It is important to point out that the continuous generation of new, latently infected cells from the active viral replication compartment generates a "continuous archive" of the changes in the virus throughout the disease, including the mutated treatment-resistant genomes, and the immune escape variants. The latent compartment is therefore not static and HIV stores its "history" here. This represents a mechanism of escape both from antiretroviral drugs and from future vaccines51.
Conclusion
Recent years have seen extremely important advances in our understanding of the immune pathogenesis of HIV infection. It can be said that AIDS immunology is finally developing along solid scientific lines. Nevertheless, there remain two huge challenges: the impossibility of eradicating the infection and the absence of therapeutic or preventive vaccines against HIV. Converting our knowledge of HIV immune pathogenesis into therapeutic developments capable of contributing to the eradication or at least better control of the infection, and designing immunogens capable of inducing immune responses to HIV which protect against infection or progression of the disease, are the main challenges in current HIV research.
Acknowledgements
Our laboratory is funded by Spanish AIDS Research Network of the Ministry of Health, "Fundación para la Investigación y la Prevención del SIDA en España" (FIPSE 3074/99, 3120/00, 36259/01, 36453/03), Ministry of Education and Scientific Research (SAF 2000/0028) and Comunidad de Madrid.

