




La producción de corticotropina (ACTH) por tumores extrahipofisarios fue descrita por Liddle en 1962. Su incidencia es de 0,1 casos por millón de habitantes y año, y es causa del 10% de los casos de síndrome de Cushing endógeno1,2. De acuerdo con las series más amplias publicadas recientemente1,3–5 el carcinoide bronquial es la causa más frecuente de secreción ectópica de ACTH (29,4%), seguido por el cáncer microcítico de pulmón (11%), los tumores neuroendocrinos gastroenteropancreáticos (9%), el carcinoma medular de tiroides (6,8%) y el carcinoide tímico (5,4%), existiendo todavía hoy un 19% de casos en los que no es posible encontrar el tumor responsable. En ocasiones, las pruebas funcionales no invasivas no permiten diferenciar entre secreción eutópica y ectópica de ACTH y ha de recurrirse al cateterismo de senos petrosos inferiores (CSPI) para determinar el origen de la secreción hormonal. A continuación, presentamos un caso de secreción ectópica de ACTH en el que la interpretación inadecuada de los resultados del CSPI condicionó una actitud terapéutica errónea.
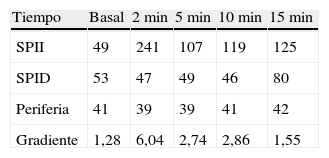
Se trata de un varón de 38 años con el único antecedente de hipertensión arterial de 2 años de evolución, bien controlada con enalapril, que consultó por primera vez en nuestro centro en noviembre de 2010. Aproximadamente 2 años antes y a causa de una infección por varicela se realizó una tomografía computadorizada (TC) en la que se objetivaron signos de osteoporosis severa, confirmada posteriormente mediante una densitometría ósea. En consulta de Reumatología se detectó cortisol libre urinario (CLU) elevado y fue remitido a Endocrinología para estudio. Clínicamente refería acúmulo graso abdominal, estrías rojo-vinosas en abdomen, muslos y axilas, debilidad de la musculatura proximal, fragilidad capilar y artromialgias generalizadas de 5 años de evolución. Se realizó un estudio funcional del eje hipófiso-adrenal que mostró los siguientes resultados: cortisol plasmático a las 8 y 23h de 27 y 23μg/dl respectivamente, CLU de 160 y 167μg/24h en 2d consecutivos, ACTH (8h) de 46 y 43pg/ml en 2d consecutivos (VN 9-55) y CLU tras supresión fuerte con dexametasona (2mg cada 6h durante 2d) de 193μg/24h. Una resonancia magnética hipofisaria no mostró hallazgos patológicos y una TC toracoabdominal evidenció una masa suprarrenal derecha de 4cm de diámetro (fig. 1). Finalmente, se realizó el CSPI que mostró un gradiente central/periferia >6 a los 2min de la administración de la hormona liberadora de corticotropina (CRH) (tabla 1). A la vista de este resultado, en enero de 2010, el paciente fue diagnosticado de enfermedad de Cushing y sometido a cirugía transesfenoidal, identificándose un microadenoma de 2mm que se resecó en su totalidad y del que no se pudo disponer de estudio histológico. Tras la cirugía el paciente no experimentó mejoría clínica y en la evaluación funcional efectuada a los 30d de la intervención se confirmó la persistencia de hipercortisolismo (CLU de 337 y 400μg/24h). En noviembre de 2010 el paciente fue remitido a nuestro centro, donde se efectuó un nuevo estudio con los siguientes resultados: cortisol plasmático a las 8 y 23h de 27 y 28μg/dl (primer día) y 26 y 26μg/dl (segundo día), CLU de 778μg/24h (VN<120; mediante quimioluminiscencia) y ACTH (8h) de 24 y 25pg/ml (VN 9-55). Tras la supresión fuerte con dexametasona el CLU apenas se modificó (720μg/24h; disminución de un 7%) y el estímulo con la CRH indujo un incremento mínimo de ACTH (basal 27; pico de 28pg/ml) y cortisol (basal 26, pico de 29μg/dl). Estos resultados condujeron al diagnóstico de síndrome de Cushing por secreción ectópica de ACTH y explicaron la ausencia de mejoría tras la cirugía hipofisaria.

Simultáneamente, el estudio funcional de la masa suprarrenal reveló unos niveles de metanefrinas urinarias 3 a 5 veces por encima del límite superior de la normalidad (metanefrinas no fraccionadas: 3,64mg/24h [VN<1], normetanefina: 2.194μg/24h [VN<440] y metanefrina: 1154μg/24h [VN<340]). Los estudios gammagráficos tanto con MIBG como con In111-octreótide mostraron únicamente depósito en la glándula suprarrenal derecha, lo que permitió confirmar el diagnóstico de feocromocitoma y excluir otras potenciales fuentes de ACTH.
En febrero de 2011 se realizó una suprarrenalectomía laparoscópica derecha previo bloqueo alfa adrenérgico. Tras la cirugía se constató una normalización de la tensión arterial y de las metanefrinas urinarias (normetanefrina: 266μg/24h, metanefrina: 104μg/24h) y un desarrollo de la insuficiencia suprarrenal (cortisol plasmático basal: 0,3μg/dl), indicativa esta última de exéresis completa de la fuente de ACTH. La pieza quirúrgica pesó 39,2g y al corte se pudo apreciar una masa bien delimitada de color pardo que comprimía el tejido glandular normal. El examen microscópico reveló amplias áreas centrales de necrosis sin atipias citológicas, imágenes de invasión vascular ni extensión extracapsular y la inmunohistoquímica fue positiva para cromogranina, S100 y EMA.
La inmunorreactividad para la ACTH (fig. 2) apoyó el diagnóstico de feocromocitoma productor de ACTH. No se encontraron mutaciones en el estudio de los genes VHL, RET y SDHB.
 Tinción con hematoxilina y eosina que muestra patrones alveolares (predominante) y trabeculares sin evidencia de necrosis tumoral confluyente, atipia citológica, invasión vascular o extensión extracapsular. (B) Inmunohistoquímica positiva para ACTH.")
El primer caso de feocromocitoma productor de ACTH fue descrito en 1942 por Neff6. Desde entonces se han publicado 65 casos más en la literatura, 2 de ellos en España7,8. En la revisión de Nijhoff9, que recoge 24 casos, la edad de presentación fue ampliamente variable (26-74 años), predominó la afectación del sexo femenino (21/24) y el tamaño tumoral osciló entre 2 y 6cm. La mayoría de los casos no mostraban síntomas de hiperproducción adrenérgica y casi todos presentaban ACTH por encima del rango normal (342±207pg/ml) y un síndrome de Cushing grave con hipopotasemia (70%) y diabetes (90%). Hasta el momento únicamente se han descrito 4 casos de parangangliomas productores de ACTH (2 en la región nasosinusal, uno cervical y uno abdominal) y no tenemos conocimiento de ningún caso maligno.
De acuerdo con la literatura, nuestro paciente presentaba una lesión de tamaño relativamente grande e HTA leve como único signo de hiperproducción adrenérgica. Por el contrario, a diferencia de la mayoría de los casos publicados, los valores de ACTH se encontraban dentro de la normalidad y presentaba un síndrome de Cushing de intensidad moderada sin diabetes, hipopotasemia ni edema.
Ante una supuesta enfermedad de Cushing, la persistencia del hipercortisolismo tras la cirugía hipofisaria, como ocurrió en nuestro paciente, obliga a reconsiderar el diagnóstico etiológico y revisar los resultados de las pruebas funcionales. En nuestro caso varios factores contribuyeron al error diagnóstico inicial: la naturaleza ACTH-dependiente del síndrome de Cushing que desvió la atención de la glándula suprarrenal, el valor del cortisol libre urinario inicial (de 160 y 167ug/d), así como la falta de supresión tras la frenación fuerte con dexametasona, lo cual orienta hacia un origen ectópico más que hipofisario, junto con la interpretación errónea del CSPI10. Esta técnica es el método más preciso para identificar el origen de la secreción de ACTH en el síndrome de Cushing ACTH-dependiente, por lo que, en caso de discrepancia, su dictamen prevalece sobre el de otros procedimientos diagnósticos. Un gradiente central/periferia ≥2:1 en una situación basal o ≥3:1 tras un estímulo con la CRH es indicativo de secreción hipofisaria de ACTH. Para su correcta interpretación debe realizarse en una situación de hipercortisolismo, de lo contrario se corre el riesgo de obtener falsos positivos derivados de la recuperación funcional de las células corticotropas y, en consecuencia, de su capacidad de respuesta a la CRH. Habitualmente se obtienen resultados concordantes en la mayoría de los tiempos analizados a lo largo de la prueba: la presencia de gradiente en un solo punto en ausencia de incidencias durante la ejecución de la misma, si bien es de difícil explicación, ha de considerarse un falso positivo y no debería condicionar la interpretación global de la prueba. No obstante, en caso de duda, es recomendable repetir el procedimiento o recurrir a otros tests diagnósticos.
En conclusión, aunque se trata de una entidad poco frecuente, la posibilidad de un feocromocitoma productor de ACTH debe ser considerada en todo paciente con síndrome de Cushing ACTH-dependiente que presente una lesión adrenal. El reconocimiento de las limitaciones de los diferentes procedimientos diagnósticos contribuye a minimizar errores diagnósticos y tratamientos inapropiados.
