


Los avances experimentados en la última década en la técnicas de estudio de los genes han supuesto una simplificación sin precedentes en el estudio de las bases genéticas de las enfermedades, acelerando el descubrimiento de genes causales o implicados en su desarrollo, y se vislumbra un horizonte en el que la secuenciación del genoma de cada ser humano será posible con rapidez y a un bajo coste. Conocemos bien las bases genéticas de los defectos en la formación de las hormonas tiroideas y conocemos parcialmente las bases genéticas de los defectos en la ontogenia tiroidea; en los últimos 4 años se han descubierto los genes causales de 2 nuevos síndromes que cursan con una disminución en la sensibilidad de la acción de las hormonas tiroideas y que afectan a su transporte (mutaciones en MCT8) y su metabolismo intracelular (mutaciones en SECISBP2); conocemos las bases genéticas de los adenomas tóxicos y bocios multinodulares tóxicos adenomatosos, y se han identificado varios genes implicados en el desarrollo de adenomas foliculares del tiroides. Sin embargo, aún no conocemos todos los genes implicados en la ontogenia tiroidea, desconocemos las bases genéticas del bocio multinodular hiperplásico, tan prevalente en algunas áreas geográficas del estado, así como de la mayoría de los trastornos tiroideos autoinmunitarios. Tenemos por delante retos importantes en cuanto a conocer las bases genéticas de los procesos tiroideos benignos que, unido a su elevada prevalencia y a la potencia de la tecnología de que disponemos o dispondremos en poco tiempo, nos ofrece un futuro prometedor y excitante en este campo de la investigación.
The advances made in the last decade in gene analysis techniques have greatly simplified the study of the genetic bases of disease, hastening identification of the genes causing or involved in disease development. Rapid and low-cost genome sequencing in all individuals may become a reality. The genetic bases of defects in thyroid hormone formation have been well defined, and those of defects in thyroid ontogeny have been partially defined; in the last 4 years, the genes responsible for 2 new syndromes causing reduced sensitivity of the action of thyroid hormone and affecting thyroid hormone transport (MCT8 mutations) and intracellular metabolism (SECISBP2 mutations) have been discovered. The genetic bases of toxic adenomas and toxic multinodular goiters have been determined and several genes involved in the development of follicular thyroid adenomas have been identified. However, not all the genes involved in thyroid ontogeny have been identified and the genetic bases of multinodular hyperplastic goiter, highly prevalent in some regions of Spain, as well as those of most autoimmune thyroid disorders, are unknown. Major challenges remain in the characterization of the genetic bases of benign thyroid processes, which, together with their high prevalence and the current and future potential of technology, suggest a promising and exciting future in this field of research.
Los avances en técnicas de amplificación de ADN, en especial la reacción en cadena de la polimerasa (PCR)1, en técnicas de secuenciación2 y en bioinformática (www.ncbi.nlm.nih.gov/genomes/static/lcenters.html) han hecho posible que en el año 2000 se lograra secuenciar el 90% del genoma humano3, 4, y que en la actualidad dispongamos del ensamblaje de varios cromosomas (www.ncbi.nlm.nih.gov/mapview/map_search.cgi). El desarrollo en paralelo de bases de datos de genes, cada vez más completas y exactas, ha permitido, mediante secuenciación directa o estudios de ligamiento, la identificación de un número cada vez mayor de genes causantes de enfermedades monogénicas, a las que la tiroidología no es ajena (OMIM: Online Mendelian Inheritance in Man, www.ncbi.nlm.nih.gov/omim/; HGMD: The Human Gene Mutation Database, http://www.hgmd.org/). Los avances en citogenética molecular5, con el desarrollo de técnicas como hibridación in situ por fluorescencia (FISH), cariotipificación espectral (SKY), FISH-multiplex (M-FISH), hibridación genómica comparativa (CGH) y CGH-arrays, han permitido el salto de la citogenética clásica, basada en la descripción de la estructura cromosómica y la identificación de aberraciones genómicas a gran escala, a una aproximación a nivel molecular, permitiendo una mayor resolución en la identificación de reordenamientos cromosómicos, el estudio de la estructura de la cromatina y su función, e incluso el estudio en tres dimensiones del genoma dentro del núcleo de células humanas intactas; avances que además han permitido el desarrollo de un número importante de bases de datos sobre variaciones estructurales de los cromosomas6. Las técnicas de ADN recombinante permiten la creación de animales transgénicos, y han posibilitado la puesta en marcha de proyectos como el NIH Knockout Mouse Project (http://www.nih.gov/science/models/mouse/knockout/index.html), que también están contribuyendo a aclarar los mecanismos por los cuales alteraciones monogénicas originan enfermedad. Los avances en el conocimiento de la regulación de la expresión de genes, incluyendo la influencia de fenómenos epigenéticos, la disponibilidad de plataformas que permiten la evaluación en un solo ensayo de la expresión de la mayoría del genoma humano7 y la introducción de técnicas de silenciamiento específico o interferencia de la transcripción de genes (siARN)8 están contribuyendo notablemente a nuestro conocimiento de las bases genéticas de las enfermedades tiroideas. En el año 2003, el National Human Genome Research Institute (NHGRI, http://www.genome.gov/), en Estados Unidos, inició un ambicioso programa conocido como ENCODE (ENCyclopedia Of DNA Elements) con la finalidad de identificar todos los elementos funcionales del genoma humano. Estos avances científicos y tecnológicos en el campo de la genética y de la genómica nos ofrecen unas expectativas insospechadas hace una década en el campo de la tiroidología. Así, desde mediados de los años noventa estamos asistiendo al descubrimiento de un número cada vez mayor de anomalías genéticas causales de enfermedades tiroideas, que comprenden desde trastornos monogénicos, que causan los trastornos del desarrollo glandular y dishormonogénesis, hasta procesos en los que están implicados varios genes, como el bocio multinodular hiperplásico o la enfermedad de Graves-Basedow.
BASES GENÉTICAS DEL HIPOTIROIDISMO CONGÉNITO NO ENDÉMICOTrastornos en la ontogenia de la glándula tiroideaEl tiroides aparece en el embrión humano a finales de la tercera semana, aunque la organización de los folículos se produce en la semana 10. Las células foliculares del tiroides proceden de un grupo de células de origen endodérmico situadas en la parte posterior de la línea media de la cavidad bucal embrionaria. En su migración descendente, las células foliculares se unen a las células C procedentes de los cuerpos ultimobranquiales de la cuarta bolsa faríngea, formando así el tiroides. A pesar de que las bases genéticas de los trastornos del desarrollo glandular son en su mayoría desconocidas, se conoce la participación de al menos 4 factores de transcripción seminales en el desarrollo y la función tiroideas: TITF1 (factor de transcripción tiroideo 1), FOXE1 (del inglés, forkhead box E1) o TITF2 (factor de transcripción tiroideo 2), PAX8 (del inglés, paired box gene 8) y HHEX (del inglés, homeobox, hematopoietically expressed).
TITF1Este gen está situado en el cromosoma 14 (14q13) y codifica TITF1, proteína de la familia de los factores de transcripción NKX2, necesaria para la supervivencia de la célula tiroidea. La expresión de TITF1 se inicia en el primordio tiroideo y persiste durante todos los estadios del desarrollo y en la etapa adulta, y también se expresa en pulmón y prosencéfalo. Ratones sin TITF1 son inviables y mueren al poco de nacer como resultado de las malformaciones pulmonares; también presentan graves anomalías cerebrales, hipofisarias y tiroideas. Hasta la fecha no se han comunicado casos de seres humanos con mutaciones homocigotas en TITF1, probablemente debido a su no viabilidad. TITF1 controla la expresión de surfactante y de la proteína morfogénica Bmp4, necesaria para el desarrollo pulmonar y causante de las alteraciones morfogénicas que aparecen en mutaciones en TITF1. También controla la expresión del factor de crecimiento de fibroblastos 8 (Fgf8) en la hipófisis, necesario para evitar la apoptosis, lo que explica la ausencia de hipófisis en los ratones knock-out para TITF1. En humanos, se han comunicado varios casos de haploinsuficiencia del gen TITF1, debido a amplias deleciones cromosómicas9 o por mutaciones heterocigotas en el gen10, 11. Entre otras manifestaciones, los sujetos afectos presentan hipotiroidismo con una glándula tiroidea de morfología normal, hipoplásica o agenesia tiroidea. La asociación de mutaciones en TITF1 con este cuadro clínico muestra su importancia en el desarrollo y la función de la glándula tiroidea.
FOXE1 o TITF2Este gen, que se localiza en el cromosoma 9q22, consta de un único exón y se expresa en la glándula tiroidea, endodermo del intestino anterior, ectodermo craneofaríngeo, paladar y bolsa de Rathke12. FOXE1 participa en la supervivencia de las células precursoras tiroideas, y mutaciones homocigotas en este gen causan agenesia tiroidea, paladar hendido, epiglotis bífida, atresia de coanas y pelo de punta13.
PAX8Se localiza en el cromosoma 2, consta de 11 exones y codifica una proteína fundamental en la diferenciación, el mantenimiento y la proliferación de tirocitos14, regulando la expresión de tiroglobulina (Tg) y tiroperoxidasa (TPO)15. En humanos, mutaciones heterocigotas en PAX8 causan hipotiroidismo congénito con hipoplasia eutópica o ectópica e incluso agenesia tiroidea16.
La tirotropina (TSH) es el principal factor trófico del tiroides, actúa a través de un receptor tipo serpentina con capacidad de activar la vía del adenosinmonofosfato cíclico (AMPc), y así regula la proliferación y la función de las células tiroideas adultas. TSHR, el gen del receptor de TSH, se localiza en el cromosoma 14q31 y consta de 10 exones; algunas mutaciones en TSHR causan resistencia a la acción de TSH y dan lugar a un espectro de manifestaciones que van desde elevaciones en la TSH con concentraciones normales de hormonas tiroideas hasta cuadros de hipotiroidismo grave debido a hipoplasia tiroidea16. La unión de la TSH a su receptor da lugar a la activación de las proteínas Gs de membrana plasmática, que a su vez generan AMPc. La subunidad alfa de la proteína heterotrimérica Gs (Gsα) está codificada por el gen GNAS, situado en el cromosoma 20q13 y formado por 13 exones; la regulación de su expresión es compleja, además de estar sometida a imprinting que es variable en función del tejido donde se exprese17. Existen mutaciones en GNAS que causan resistencia a la acción de la TSH cuando el alelo mutado proviene de la madre, lo que obedece al hecho de que en el tiroides hay imprinting paterno (se anula el alelo paterno) y se expresa de forma preferencial el alelo materno18.
Defectos en la formación de las hormonas tiroideasLos defectos en la hormonogénesis representan el 10% de los casos de hipotiroidismo congénito y sus bases genéticas están bien definidas. Estos trastornos se heredan de forma autosómica recesiva, y la mayoría presenta bocio debido al aumento en las concentraciones séricas de la TSH en respuesta a un descenso en los valores de hormonas tiroideas.
SLC5A (salute carrier family 5, member 5) es el gen que codifica el transportador de yodo NIS (del inglés, sodium/iodide symporter), se localiza en el cromosoma 9 y consta de 15 exones. NIS se expresa en tiroides, glándula salival, mucosa gástrica y plexos coroideos. Mutaciones en SLC5A5 dan lugar a hipotiroidismo congénito con bocio, en ocasiones de grandes proporciones, con escasa o nula captación de yodo radiactivo19.
TPO es el gen que codifica TPO, enzima que cataliza las reacciones de oxidación y organificación que dan lugar a la unión covalente entre yodo y tirosina para formar yodotirosinas, y el acoplamiento de éstas para formar T3 y T4. TPO se localiza en el cromosoma 2p25, está compuesto por 17 exones y codifica una proteína de 933 aa, con un lugar de unión al grupo heme, localizada en el borde apical del tirocito. La deficiencia de TPO es la causa más frecuente de hipotirodismo congénito dishormonogénico, que causa defectos de organificación total14. La mayoría de las mutaciones se localizan en los exones 8 a 10, donde se codifican las histidinas proximales y distales al grupo heme, la parte funcional de TPO.
Tg es el gen que codifica la proteína Tg de síntesis exclusiva en el tiroides, se localiza en el cromosoma 8q24, consta de 48 exones y una región codificante que supera los 8.244bp. El monómero de Tg consta de 2.749 aminoácidos y contiene 66 tirosinas, lugares donde se incorpora el yodo para formar la monoyodotirosina (MIT) y la diyodotirosina (DIT) mediante un proceso químico que requiere de TPO y H2O2. Hasta la fecha se han descrito 35 mutaciones, que comprenden sustituciones, deleciones de exones e introducción de codones de parada prematuros20, y causan un cuadro de hipotirodismo congénito con intensidad variable y bocio con concentraciones séricas de Tg bajas. La asociación de mutaciones monoalélicas en Tg con bocio simple es objeto de controversia21.
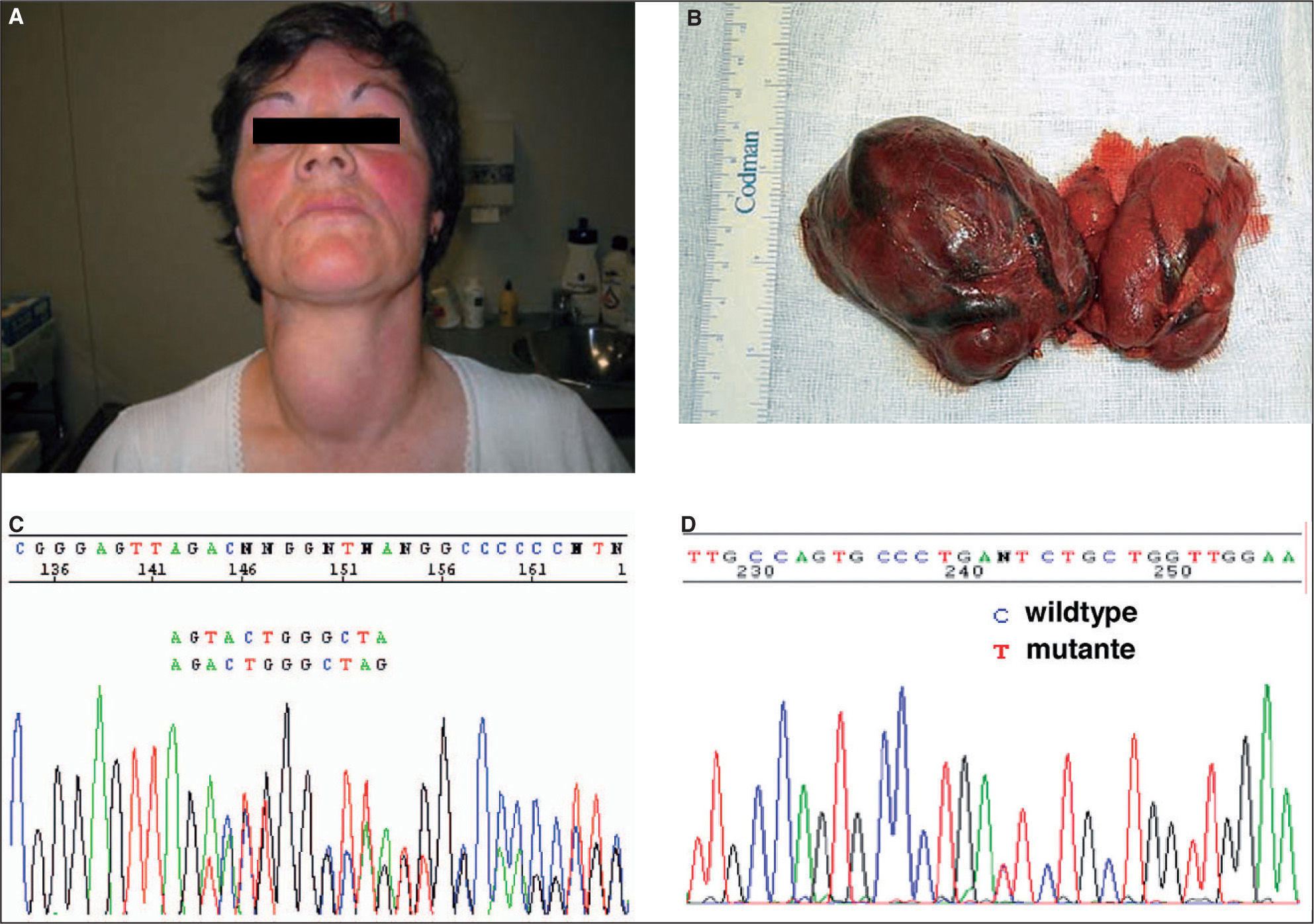
SLC26A4 (pendrina o solute carrier family 26, member 4) se localiza en el cromosoma 7q31 y codifica pendrina, proteína transmembrana que se expresa en tiroides, oído interno, endometrio y riñón. En el tiroides se localiza en la membrana apical y participa en el transporte de yodo desde el interior de la célula al coloide. Mutaciones en SLC26A4 causan síndrome de Pendred, cuadro autosómico recesivo caracterizado por sordera neurosensorial y bocio asociado con frecuencia a hipotiroidismo (fig. 1). SLC26A4 también causa sordera recesiva neurosensorial no síndrómica 4 o DFNB4 (http://www.medicine.uiowa.edu/pendredandbor/slcMutations.htm).
 a expensas sobre todo del lóbulo tiroideo derecho en donde había varios nódulos tiroideos grandes (B). La paciente es portadora de dos mutaciones diferentes (heterocigota compuesta) en el gen SLC26A4, c.279delT que causa una alteración en la pauta de lectura en el exón 3 (C), y c.578C>T en el exón 5 (D).")
Paciente de 43 años con sordera y bocio (A) a expensas sobre todo del lóbulo tiroideo derecho en donde había varios nódulos tiroideos grandes (B). La paciente es portadora de dos mutaciones diferentes (heterocigota compuesta) en el gen SLC26A4, c.279delT que causa una alteración en la pauta de lectura en el exón 3 (C), y c.578C>T en el exón 5 (D).
DUOX1 (THOX1), o dual oxidasa 1, y DUOX2 (THOX2), o dual oxidasa 2, son genes localizados en el cromosoma 15q15.3 y formados por 35 y 34 exones, respectivamente, que codifican NADPH oxidasas implicadas en la producción de H2O2. Pacientes con mutaciones homocigotas o heterocigotas compuestas en THOX2 presentan un defecto en la organificación de yodo que origina hipotirodismo congénito grave sin bocio22, mientras que pacientes con mutaciones en un solo alelo presentan un hipotiroidismo neonatal transitorio que, sin embargo, puede ser grave22.
BASES GENÉTICAS DE LOS SÍNDROMES DE DISMINUCIÓN EN LA SENSIBILIDAD A LA ACCIÓN DE LAS HORMONAS TIROIDEASLos síndromes de disminución en la sensibilidad a la acción de las hormonas tiroideas son trastornos genéticos causados por una disminución en la respuesta de órganos y tejidos diana a las hormonas tiroideas, cuyo origen radica en cualquiera de los pasos desde que las hormonas tiroideas penetran la membrana plasmática hasta que su unen a sus receptores nucleares.
Mutaciones en el transportador de monocarboxilatos 8 (MCT8)Las hormonas tiroideas penetran a la célula a través de transportadores de membrana de mayor o menor especificidad23, 24. MCT8 es un transportador específico de hormonas tiroideas codificado por el gen SLC16A2 (solute carrier family 16, member 2), localizado en el cromosoma Xq13.2 y que consta de 6 exones. MCT8 tiene 539 aminoácidos y 12 dominios transmembrana, con la peculiaridad que las regiones N-terminal y C-terminal se localizan a nivel intracelular. Su expresión es ubicua; el hígado es el órgano con mayores grados de expresión aunque es en el área neuronal donde su papel es especialmente relevante, ya que al carecer éstas de capacidad para la síntesis de T3, la incorporación de la hormona es a partir de los astrocitos, utilizando a MCT8 como transportador específico. Mutaciones en MCT8 originan un cuadro24, 25 ligado al cromosoma X, que afecta a varones hemocigotos y que produce retraso psicomotor grave, hipotonía y cuadriplejía espástica. En cuanto a la bioquímica, hay elevación de la T3 sérica, disminución de T4 total y libre y la reversa del T3, y TSH normal o elevada. Las madres portadoras presentan fenotipo bioquímico pero no clínico, lo que indica inactivación específica del alelo mutado en el tejido nervioso, o que las mujeres expresan los dos alelos sin que se produzca inactivación de la parte de cromosoma X donde se localiza SLC16A2; entonces el fenotipo bioquímico es consecuencia de haploinsuficiencia25.
Mutaciones en SECISBP2 (SECIS binding protein 2)Las desyodinasas son selenoproteínas que catalizan las desyodinación de las yodotironinas, por lo que intervienen en la activación y la inactivación de las hormonas tiroideas. Para ello, necesitan la incorporación de selenio a las selenocisteínas de su centro activo. La incorporación de selenocisteína requiere el reconocimiento en el ARN mensajero que codifican las selenoproteínas, de una secuencia de incorporación de selenocisteína (SECIS) a través de un complejo del que forma parte la SECISBP2. El gen de SECISBP2 se localiza en el cromosoma 9q22.2, consta de 17 exones y codifica una proteína de 854 aminoácidos. Mutaciones recesivas26 en SECISBP2 causan un fenotipo tiroideo similar al observado en ratones knock-out para desyodinasas 1 y 2, que consiste en elevaciones de T4, reversa de T3 y TSH sérica con concentraciones bajas de T3. Este fenotipo es consecuencia de una menor actividad de las desyodinasas inducidas por un defecto en la incorporación de selenio secundario a las mutaciones en SECISBP2.
Mutaciones en el receptor beta de las hormonas tiroideasLos receptores de las hormonas tiroideas son factores de transcripción nucleares conocidos como receptor beta (TRβ) y receptor alfa (TRα); a su vez, hay 2 isoformas de cada uno de ellos, TRβ1 y TRβ2 y TRα1 y TRα2. THRB, el gen del TRβ, se localiza en el cromosoma 3 y consta de 10 exones, mientras que el gen del TRα, THRA, se localiza en el cromosoma 17 y lo forman 9 exones. Mutaciones en THRB causan el síndrome de resistencia a la acción de las hormonas tiroideas (SRHT); el 75% de los casos son familiares y heredados de forma autosómica dominante; el 15%, casos esporádicos, causados por mutaciones de novo, y en el resto de los casos se desconoce la localización de las mutaciones causales. La mayoría de las mutaciones en TRHB son sustituciones; en las proteínas se traducen en el cambio de un aminoácido por otro con la subsecuente modificación de la estructura y la función del receptor. Estas mutaciones se localizan en la parte de unión a la T3, lo que impide que la hormona se una al receptor, y no se han descrito mutaciones en los dominios de unión al ADN23. El TRβ mutado actúa de forma dominante negativa, debido a la competición entre el heterodímero RXR-TRβ mutado con el normal por la unión al ADN; al no tener el heterodímero mutado capacidad de unión a la T3, no puede liberar los correpresores unidos al complejo y reprime así la transcripción basal de forma permanente27.
BASES GENÉTICAS DE LAS ENFERMEDADES TIROIDEAS AUTOINMUNITARIASLa enfermedad de Graves (EG) y la tiroiditis de Hashimoto (TH) representan el 30% de todas las enfermedades autoinmunitarias organoespecíficas. Estudios en gemelos monocigotos y estudios de segregación apuntan a una base hereditaria en la susceptibilidad a presentar una enfermedad tiroidea autoinmunitaria28. Mediante estudios de ligamiento y asociación, análisis de genes candidatos y cribado masivo del genoma a través de microsatétiles y SNP, se han identificado 7 locus potencialmente implicados29, 3 ligados a EG y TH (6p, 8q, 10q), 3 ligados a EG (7q, 14q, 20q) y 1 locus ligado a HT (12q), así como unos pocos genes que confieren mayor susceptibilidad para presentar una enfermedad tiroidea autoinmunitaria:
- 1.
Genes reguladores de la inmunidad. Aunque la alteración genética primaria se desconoce, los genes del complejo mayor de histocompatibilidad (CMH) de la clase II, localizados en el cromosoma 6p21, son un componente principal en la predisposición genética a las enfermedades tiroideas autoinmunitarias. CTLA-4 (cytotoxic T-lymphocyte-associated protein 4), situado en el cromosoma 2q33, codifica el receptor CTLA-4, homólogo a CD28, sobre el que actúan moléculas B7 y que se expresa en linfocitos T CD4+ y CD8+ recién activados; su función es inhibir la activación de linfocitos T y así contrarrestrar las señales liberadas por CD28. Las moléculas del CMH de clase II y CTLA-4 confieren una susceptibilidad del 50% de presentar enfermedad tiroidea autoinmunitaria.
- 2.
Genes específicos del tiroides. Estudios de ligamiento y asociación han mostrado una relación significativa entre varios polimorfismos del gen Tg30 y la enfermedad tiroidea autoinmunitaria, en especial con el polimorfismo Tgms2 (repeticiones CA), localizado en el intrón 27, y un SNP del exón 33. La asociación con polimorfismos en TSHR y TPO está en controversia, aunque la mayoría de los datos actuales no apoyan dicha asociación.
- 3.
Síndromes poliglandulares autoinmunitarios (APS). Las enfermedades tiroideas autoinmunitarias forman parte de los APS. En el 2-13% de los casos de APS tipo I (poliendocrinopatía autoinmunitaria, candidiasis mucocutánea y distrofia ectodérmica -APECED-) existen anticuerpos antitiroideos y, en general, este cuadro está causado por mutaciones en el gen AIRE (autoinmune regulator), localizado en el cromosoma 21q22.3, formado por 14 exones, que codifica una proteína de 545 aminoácidos, cuya función es actuar como un factor de transcripción en el timo. Sin embargo, su participación en la EG y la TH que se presentan de forma aislada no parece relevante31. El APS tipo II se hereda de forma poligénica y la enfermedad tiroidea autoinmunitaria se presenta en el 70% de los casos, en que se asocia a moléculas CMH clase II.
El déficit de yodo es el principal factor etiológico de bocio endémico eutiroideo, aunque estudios en familias y gemelos con bocio indican un fuerte componente hereditario y existen evidencias de transmisión autosómica dominante y ligada al cromosoma X. En estudios realizados sobre familias con varios miembros afectos de bocio multinodular, se encontraron evidencias de ligamiento en el cromosoma 14q (locus MNG1 o multinodular goiter 1)32, el cromosoma Xp22 (locus MNG2 o multinodular goiter 2)33 y el cromosoma 3q26.1-26.3 (locus MNG3 o multinodular goiter 3)34. En un estudio35 realizado sobre muestras de familias con bocio eutiroideo procedentes de Dinamarca, Alemania y Eslovaquia, en las que se analizaron 450 microsatélites, se encontró una asociación en el 20% de los casos con locus en los cromosomas 2q, 3p, 7q y 8p.
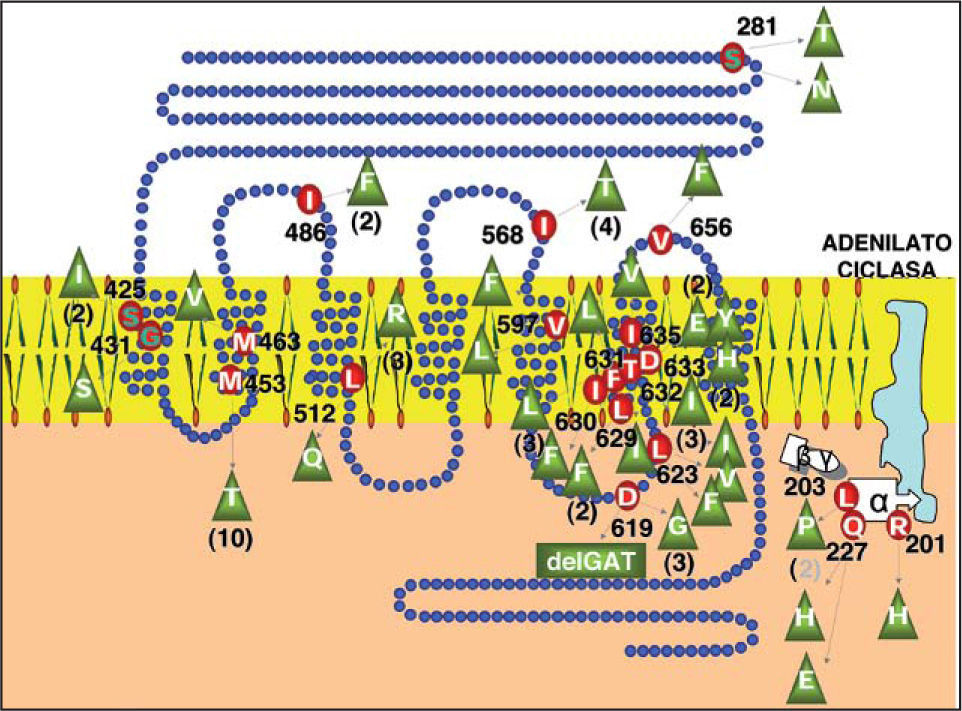
Adenoma tiroideo tóxico y bocio multinodular adenomatoso tóxicoLos adenomas tóxicos y bocios multinodulares adenomatosos tóxicos son neoplasias benignas bien encapsuladas de origen monoclonal, con función y crecimiento autónomos36. En su mayoría están causadas por mutaciones en los genes TSHR37 y/o GNAS, que dan lugar a la activación constitutiva de la vía del Amp cíclico, estimulando la función y la proliferación de los tirocitos portadores de dichas mutaciones. Dentro del TSHR la mayoría de las mutaciones residen en los exones 9 y 10, y en especial en la parte codificante del segmento 6 transmembrana (aminoácidos 619 a 633). Las mutaciones activadoras de Gsα se localizan en los codones 201 (exón 8) y codón 227 (exón 9) (fig. 2). Contrario a los hallazgos en bocios adenomatosos, en los bocios multinodulares tóxicos hiperplásicos no se ha encontrado mutaciones activadoras en TSHR y GNAS y, aunque su desarrollo está en relación con la deficiencia de yodo y selenio, sus bases genéticas se desconocen.

Distribución de mutaciones activadoras de la vía del adenosinmonofosfato cíclico en el receptor de TSH y en la subunidad alfa de la proteína Gs de membrana, en 85 adenomas tóxicos procedentes de la Comunidad Autónoma de Galicia. D: ácido aspártico; E: ácido glutámico; F: fenilalanina; G: glicina; H: histidina; I: isoleucina; L: leucina; M: metionina; Q: glutamina; R: arginina; S: serina; T: treonina; V: valina.
Las investigaciones en genética de las enfermedades tiroideas están permitiendo aclarar las bases moleculares de procesos "clásicos" como los cuadros de hipotirodismo congénito o bocios tóxicos, el descubrimiento de nuevos síndromes, como los nuevos casos de disminución en la sensibilidad a la acción de las hormonas tiroideas, y progresar al mismo tiempo en la comprensión de los mecanismos fisiológicos por los cuales las hormonas tiroideas ejercen sus acciones. El conocimiento de las bases genéticas de las enfermedades tiroideas repercute no sólo en el diagnóstico etiológico de estos procesos, sino que está permitiendo el diseño de fármacos capaces de modificar los efectos de algunas de estas mutaciones en las células del organismo. La simplificación y el abaratamiento de nuevas tecnologías aplicadas a la genética y a la epigenética se traducirán pronto en un mayor conocimiento de las bases genéticas de los procesos tiroideos.
