




Las displasias esqueléticas que cursan con alteraciones en la mineralización son difíciles de diferenciar desde el punto de vista ecográfico, debido a que comparten características comunes unas con otras. Es importante conocer el diagnóstico exacto de estas entidades para realizar un asesoramiento adecuado. El uso de la ecografía tridimensional (3D), la navegación multiplanar y las reconstrucciones nos permite visualizar mejor ciertas características ecográficas de este tipo de alteraciones, que nos puede ayudar en el diagnóstico diferencial.
Se presentan 2 casos de fetos afectos de displasias esqueléticas letales con alteración en la mineralización ósea diagnosticados prenatalmente y su dificultad en el diagnóstico.
Skeletal dysplasias with hypomineralisation are difficult to distinguish by ultrasound because they share common features. It is very important to determine the exact diagnosis and provide appropriate counselling to parents for future pregnancies. The use of 3D ultrasound, multiplanar navigation and reconstructions allows better visualization of certain ultrasonographic features of these types of alteration, which can help to establish the differential diagnosis. We report the cases of two foetuses with skeletal dysplasias and hypomineralisation and the difficulties of the differential diagnosis.
Las displasias esqueléticas son un grupo heterogéneo de anomalías genéticas raras que afectan a 2,4-4,5 de 10.000 recién nacidos1–6.
Estas anomalías, consideradas globalmente, suelen acompañarse de hallazgos ecográficos llamativos que las hacen fácilmente diagnosticables mediante ecografía. Sin embargo, al ser entidades relativamente poco frecuentes, y compartir hallazgos ecográficos muchas de ellas, su diagnóstico histológico o identificación precisa puede resultar muy difícil.
Las displasias esqueléticas que cursan con hipomineralización cursan con trastornos severos del desarrollo del hueso y cartílago y, generalmente, son difíciles de diferenciar desde el punto de vista clínico-radiológico. Es necesario realizar estudios anatomopatológicos y genéticos que pueden ser complejos y costosos para poder conocer el diagnóstico exacto y proporcionan un asesoramiento adecuado a los progenitores para futuras gestaciones.
Las displasias esqueléticas más frecuentes, y que cursan con alteraciones en la mineralización son: la acondrogénesis, hipocondrogénesis tipo i, la osteogénesis imperfecta tipo ii y la hipofosfatasia. Hoy en día el uso de la ecografía tridimensional, la navegación multiplanar y las reconstrucciones nos permite visualizar las extremidades, los rasgos faciales y ciertas características ecográficas de este tipo de alteraciones, que nos puede ayudar en el diagnóstico diferencial.
Se presentan 2 casos de fetos afectos de la mineralización ósea diagnosticados prenatalmente y su dificultad en el diagnóstico diferencial.
Caso 1Se trata de una gestante de 42 años, sin antecedentes de interés, sin historia de consanguinidad, con 3 cesáreas previas y 3 hijos sanos. Acude a la unidad de diagnóstico prenatal en la semana 26 con la sospecha ecográfica de displasia esquelética. La paciente no se había realizado ecografías previas hasta ese momento.
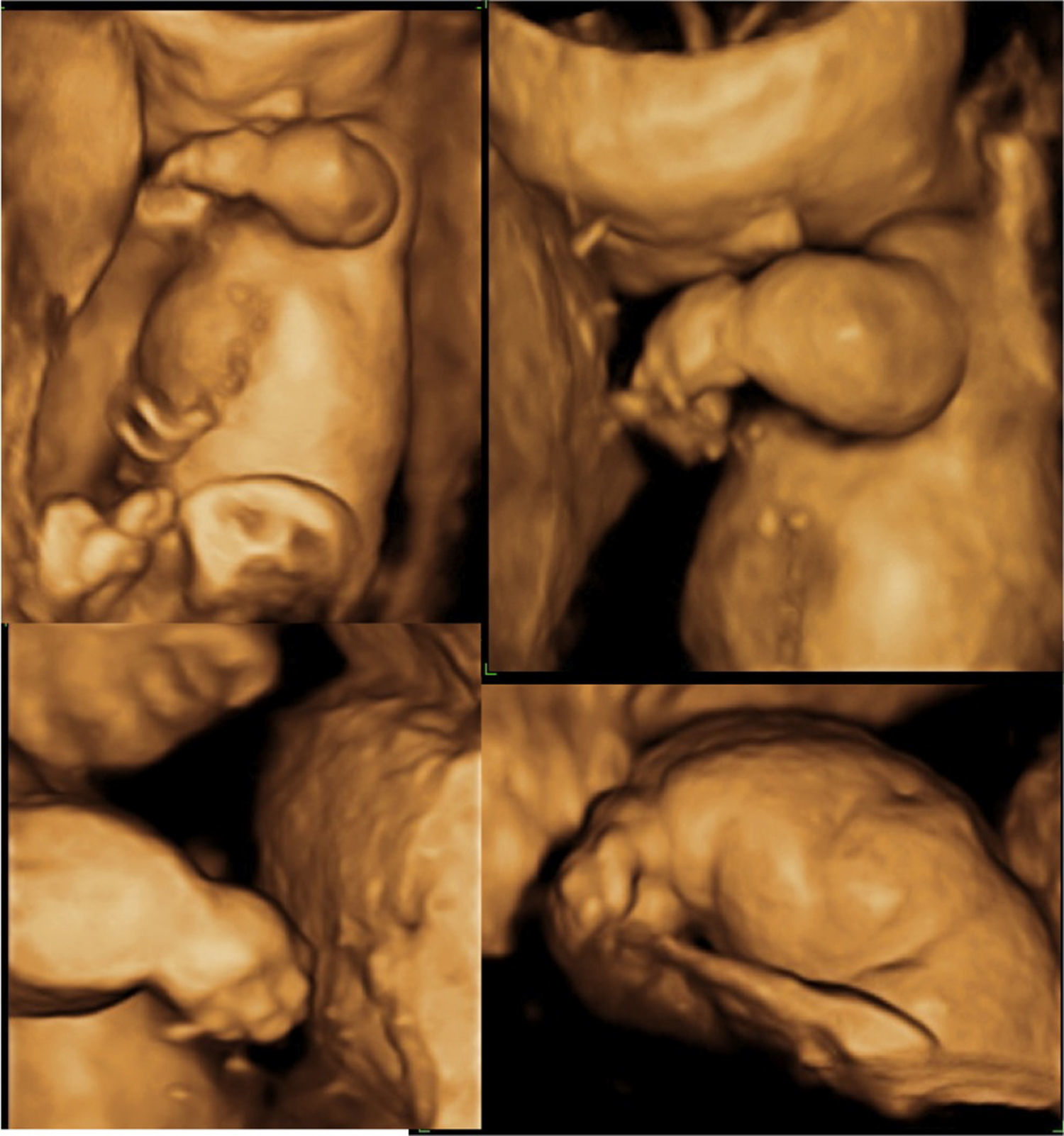
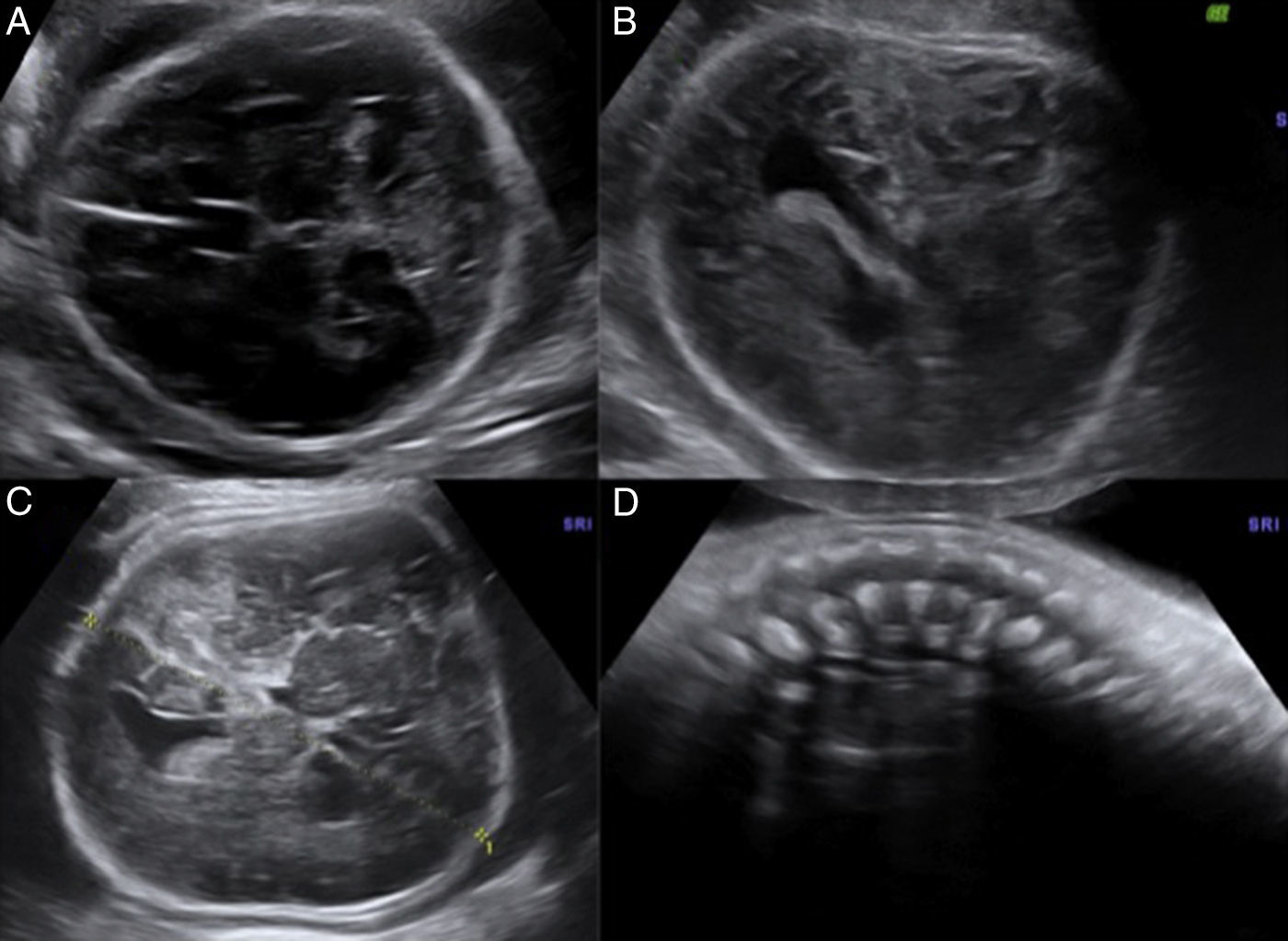
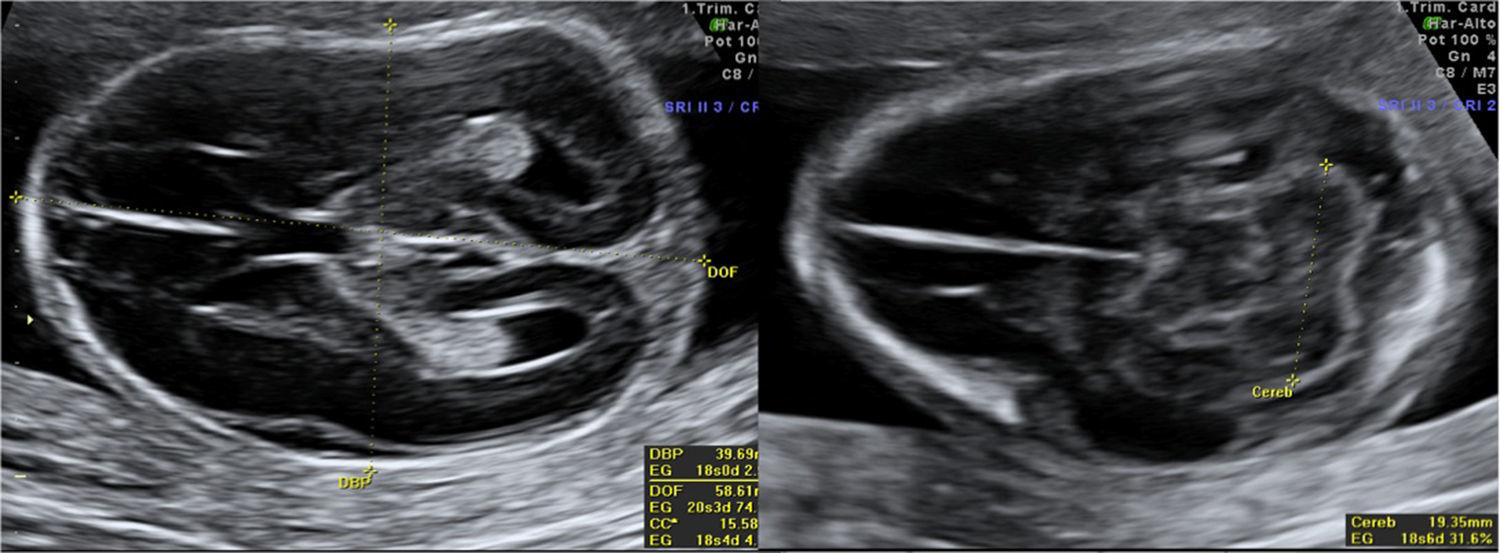
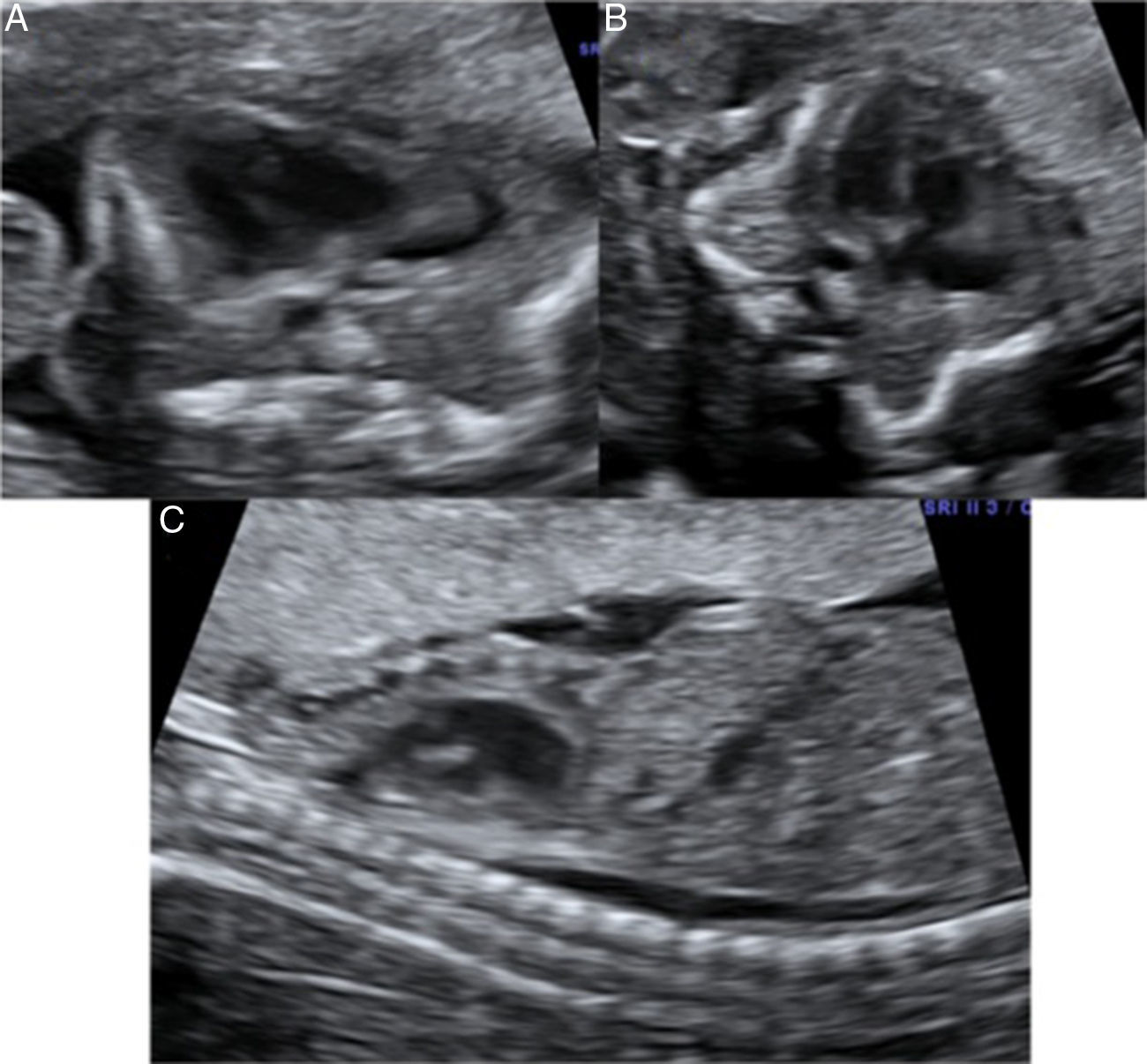
En el estudio ecográfico se observó una micromelia severa (fig. 1), y mediante reconstrucción 3D se visualizó una anomalía en las manos, con dedos juntos y pequeños, de aspecto edematoso y con posición anormal. La calota fetal presentaba una morfología anormal, con traslucidez ecográfica que permitía una visualización de las estructuras encefálicas de ambos lados con claridad, y que a la presión con el transductor se producía una depresión del hueso, hallazgos que traducen una hipomineralización ósea. Las costillas y la columna vertebral también presentaban disminución de la osificación (fig. 2).


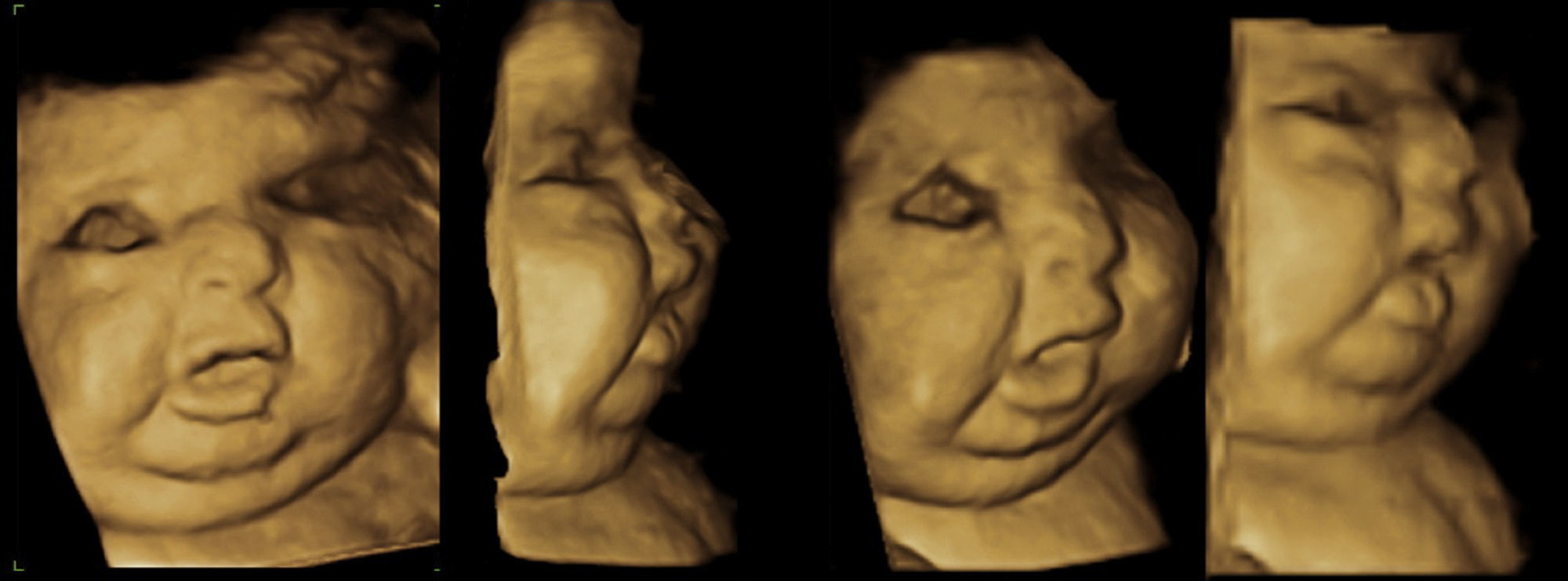
En la valoración del perfil fetal presentaba una micrognatia. Mediante la reconstrucción 3D se visualizó además otros rasgos de la cara, como un pliegue debajo del mentón y un cuello corto, la cara plana, el puente nasal deprimido (fig. 3).

El tórax era hipoplásico, las costillas hipoecogénicas y presentaba una cardiomegalia relativa (fig. 4)

Dados los hallazgos se establece el diagnóstico de displasia esquelética con alteración de la mineralización, siendo lo más probable un caso de acondrogénesis tipo i. La paciente decidió continuar con la gestación. En la semana 32 se objetiva un polihidramnios severo que requirió de 2 amniodrenajes por sintomatología respiratoria de la paciente, realizándose estudio del cariotipo en líquido amniótico siendo 46, XY normal, y estudio de acondroplasia que fue negativo. En nuestro centro no se dispone de estudios de array prenatal, por lo que no se realizó.
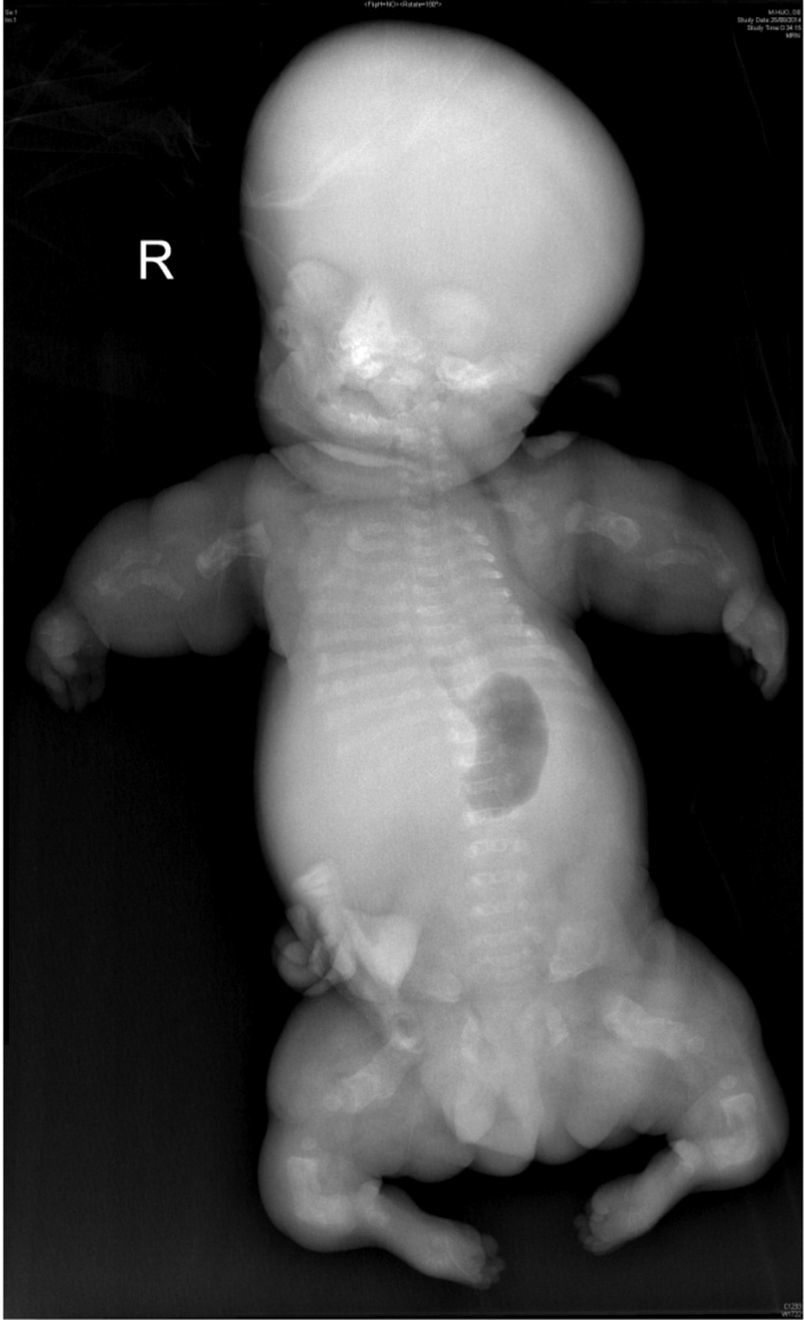
Dado que era un feto inviable se permitió un intento de parto vía vaginal, aunque la gestante tenía 3 cesáreas previas. Se produjo un parto eutócico sin incidencias en la semana 39+3, naciendo un varón que pesó 1.935g, y falleció a los 30min. Se realizó radiografía simple al feto (fig. 5) visualizándose una micromelia severa con hipomineralización en la columna lumbar predominantemente, el sacro, el pubis y el isquion; el tórax era anormal, pequeño, con forma de campana.

Radiografía simple posmortem: osificación vertebral deficiente con hipomineralización de huesos del sacro, pubis e isquion, huesos largos acortados y en ambos extremos del húmero hay ensanchamiento metafisario. Tórax estrecho con costillas cortas hipomineralizadas y con fracturas.
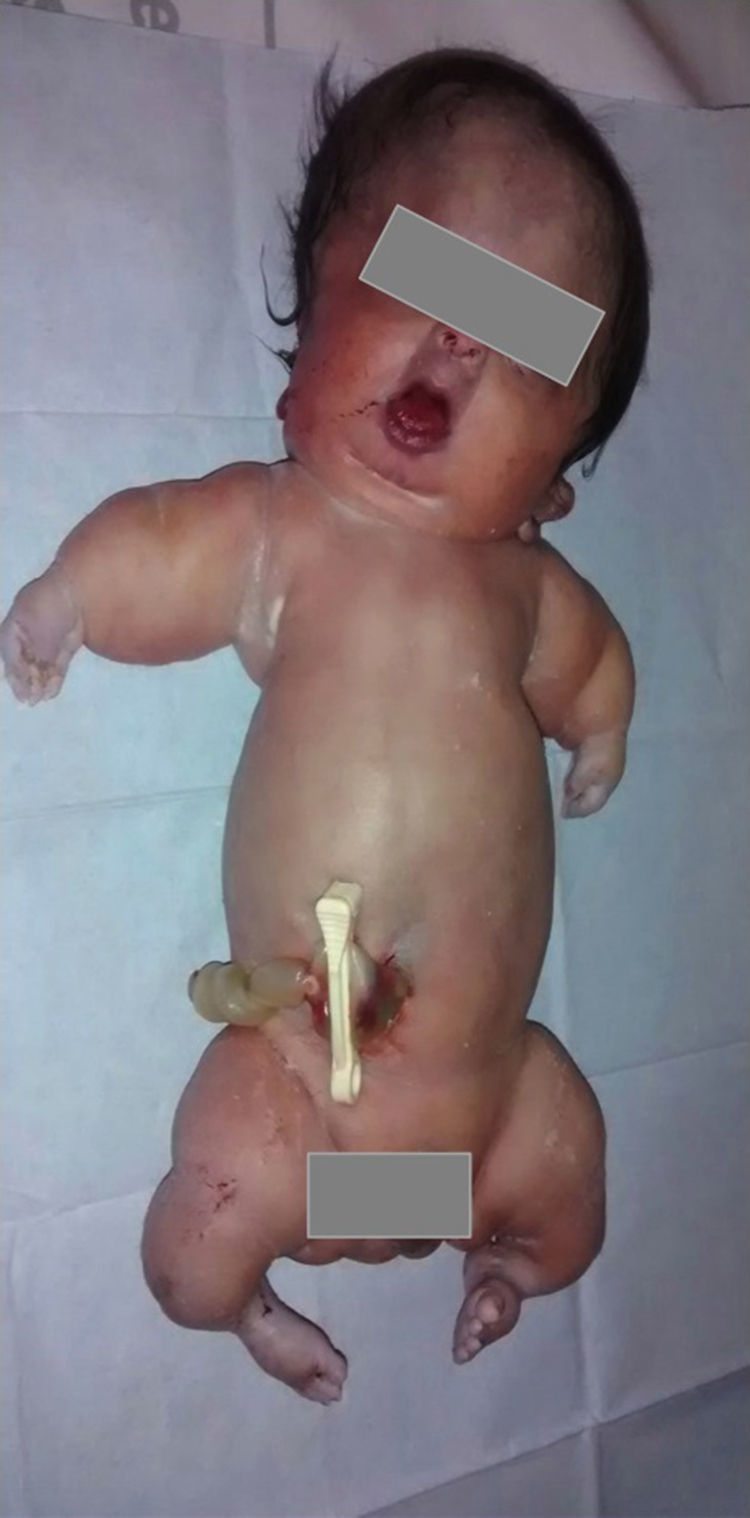
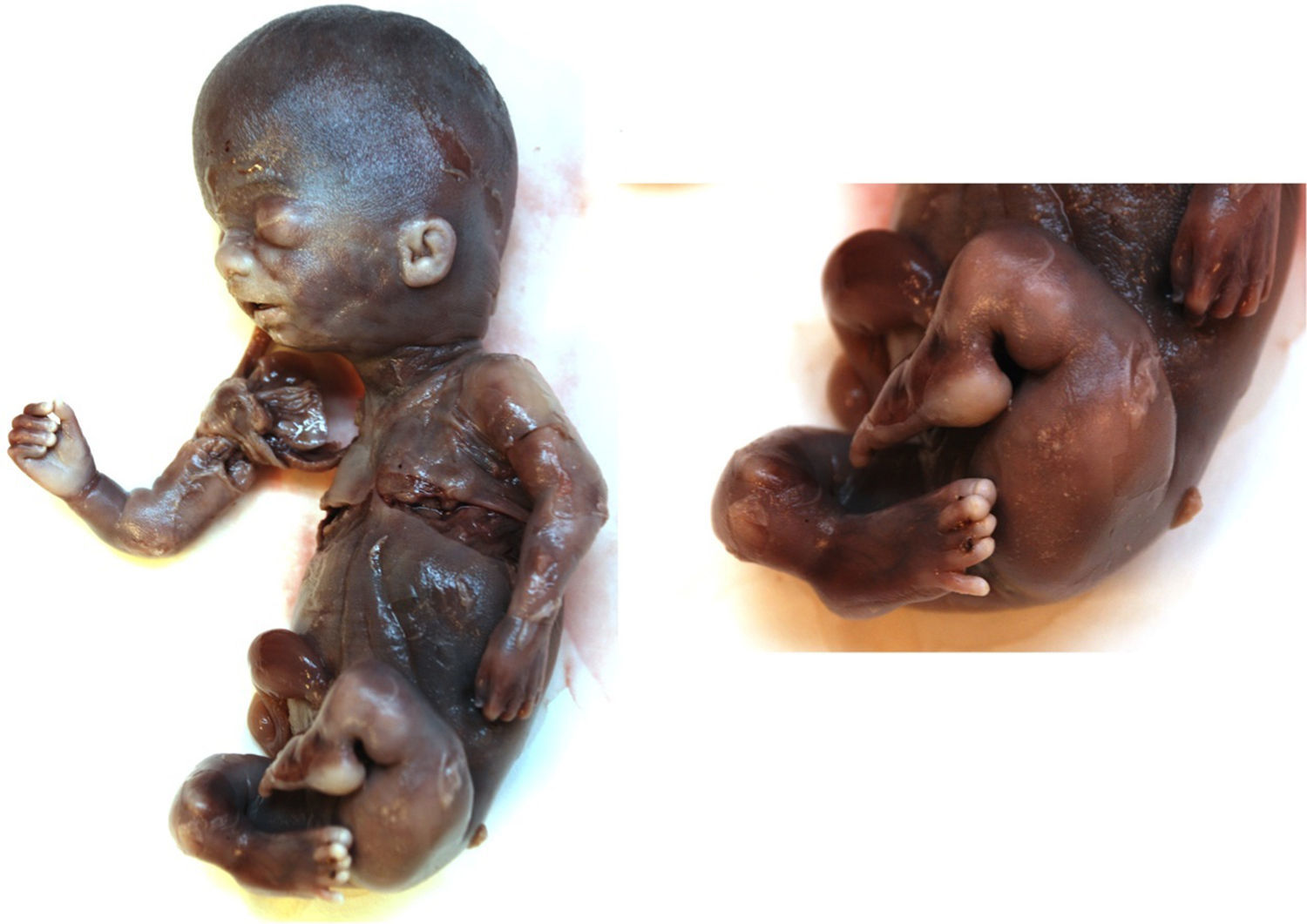
Aunque se intentó realizar un diagnóstico genético diferencial, mediante la biopsia cutánea, la contaminación de la muestra tomada impidió el aislamiento del ADN genómico. Sin embargo, en el estudio macroscópico fetal se visualizó una cabeza grande con fascies anormal, como en la reconstrucción 3D, y micromelia severa con braquidactilia (fig. 6). No se hizo estudio histopatológico por deseo de la paciente.
Caso 2
Se trata de una secundigesta de 35 años con un embarazo y parto eutócico anterior, sin incidencias, sin antecedentes familiares ni personales de interés, sin historia de consanguinidad, con gestación conseguida por técnicas de reproducción asistida (fecundación in vitro), que es remitida en la semana 19+5 por sospecha de displasia esquelética.
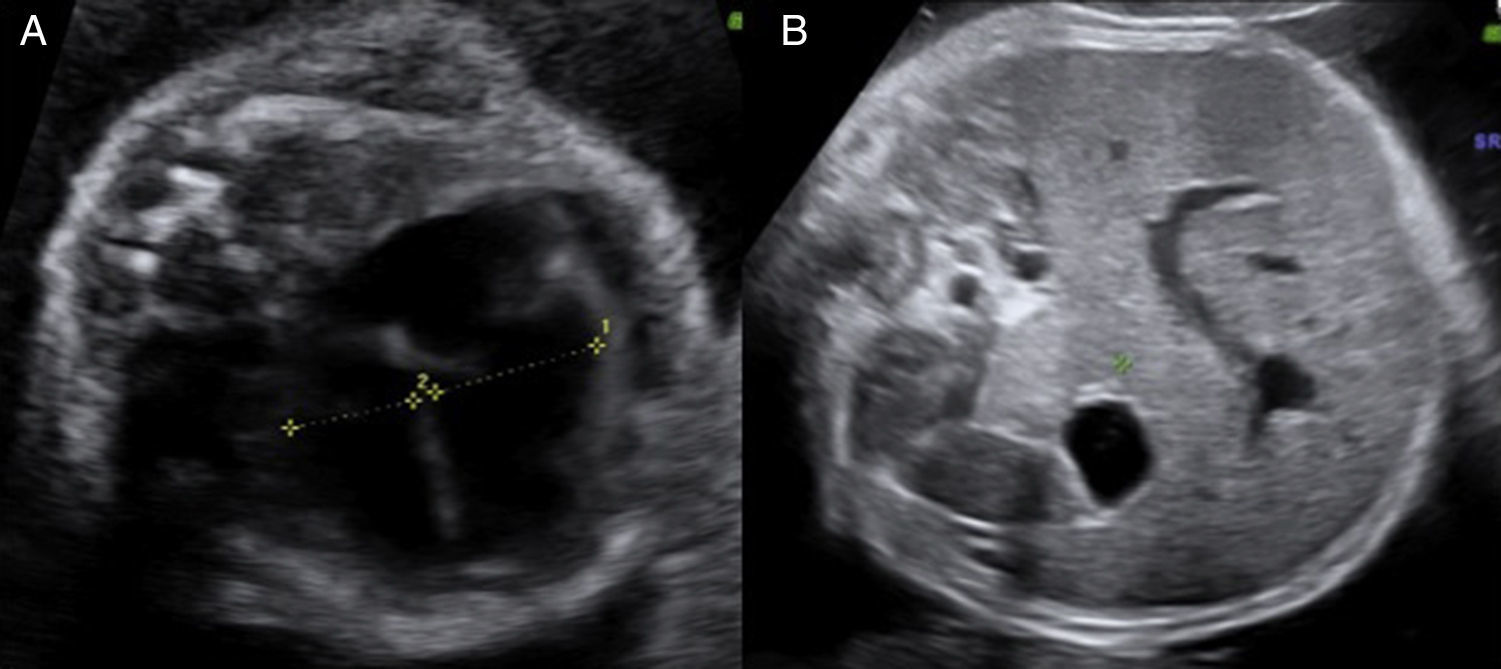
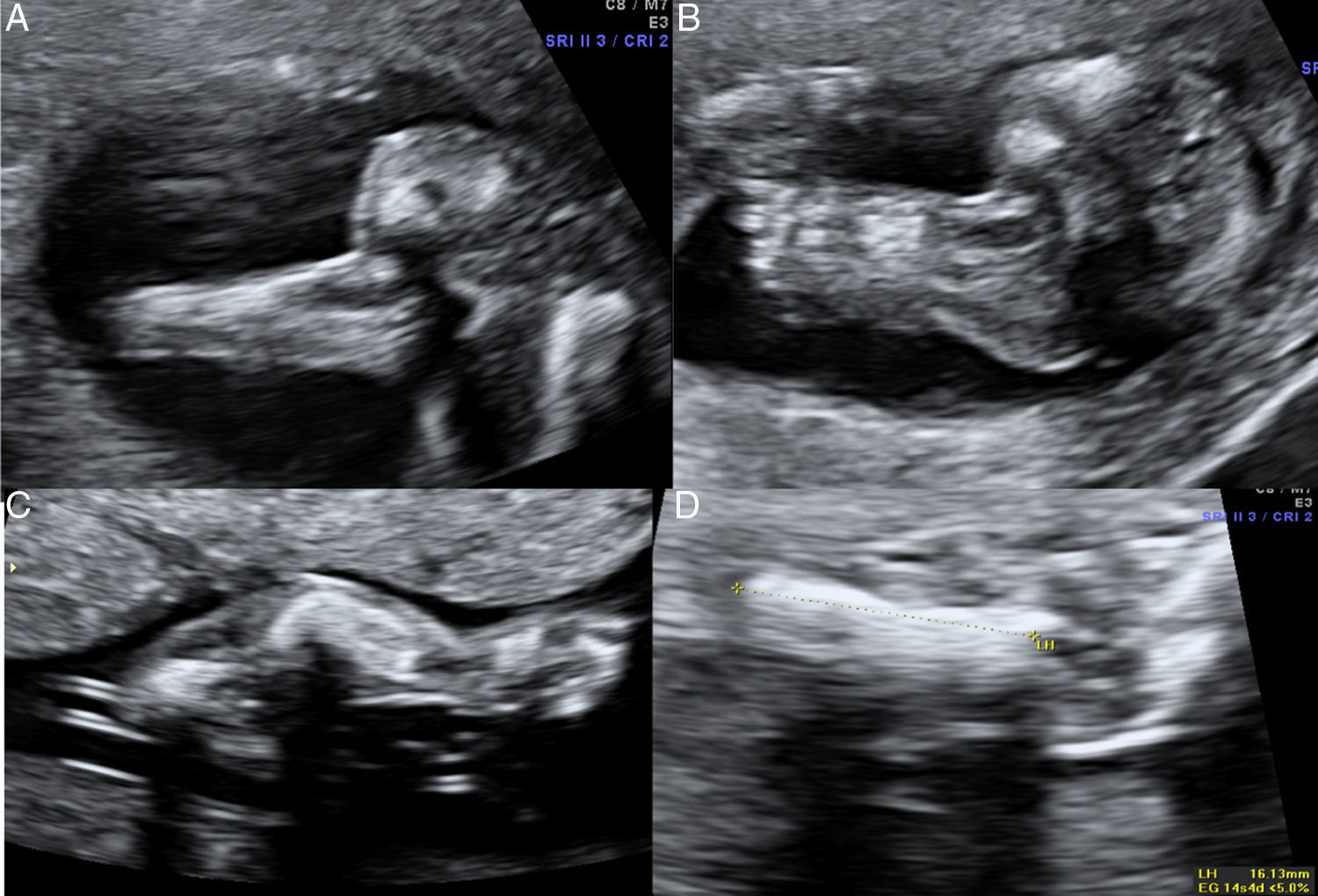
En el estudio ecográfico fetal se visualizó un feto de sexo femenino que presentaba una micromielia severa (fig. 7), a expensas de un acortamiento grave de todos los huesos largos. Además presentaba una hipomineralización de la calota fetal, que se veía tanto en el corte medio sagital como en los cortes axiales, objetivándose los huesos de la calota finos, que permiten una clara visualización de las estructuras encefálicas y que al presionar con el transductor sobre la misma se deprimen (fig. 8). El tórax estaba hipoplásico, con una cardiomegalia relativa y desproporción toracoabdominal, con ratio de 0,55 (fig. 9).


 Corte axial del tórax fetal, se visualiza un tórax pequeño con caja torácica estrecha y corazón de tamaño normal pero que impresiona de mayor tamaño por lo que se diagnostica de hipoplasia torácica con cardiomegalia relativa. C) Corte longitudinal del tórax fetal. Se objetiva un tórax pequeño en relación con el abdomen fetal.")
A y B) Corte axial del tórax fetal, se visualiza un tórax pequeño con caja torácica estrecha y corazón de tamaño normal pero que impresiona de mayor tamaño por lo que se diagnostica de hipoplasia torácica con cardiomegalia relativa. C) Corte longitudinal del tórax fetal. Se objetiva un tórax pequeño en relación con el abdomen fetal.
Dados los hallazgos ecográficos se establece como sospecha diagnóstica una acondrogénesis tipo 1A, y dado el pronóstico letal de esta enfermedad se decide la interrupción de la gestación. En el estudio anatomopatológico no se filia el tipo de displasia esquelética. En el examen macroscópico se objetivan múltiples fracturas, predominantemente en las extremidades inferiores. Llamaba la atención que el acortamiento de los huesos no era tan severo como se había visto en la ecografía, sino que estaban acortados por las fracturas que presentaban, por lo que se pensó que pudiera tratarse de una osteogénesis imperfecta tipoii (fig. 10).
Discusión
Las displasias esqueléticas con alteración en la mineralización son entidades poco frecuentes. Se estima que representan el 5% de las anomalías genéticas en el periodo neonatal4. La hipomineralización se produce por alteraciones genéticas en las proteínas que intervienen en la formación ósea. Las anomalías genéticas que afectan el desarrollo del sistema esquelético son muy numerosas. En el año 2010 se realizó una revisión de los diferentes tipos de displasias esqueléticas u osteocondrodisplasias en las que se incluyeron 456 y se agruparon en 40 grupos, según criterios moleculares, bioquímicos y/o radiológicos. De ellos 316 se asociaron con mutaciones de uno o más de 226 genes diferentes5. El tipo de herencia de las displasias esqueléticas puede ser autosómica dominante, recesiva, ligada al X dominante o recesiva y ligada al Y5. La mayoría de estas anomalías se originan de nuevas mutaciones dominantes, y las autosómicas recesivas se producen en familias sin antecedentes de displasias esqueléticas6. Es importante conocer el modo para aconsejar para futuras gestaciones, y además debe indagarse en la historia familiar para ayudarnos en el diagnóstico.
Desde el punto de vista ecográfico, en el caso de una sospecha de displasia esquelética debe: medirse todos los huesos largos, clasificar el acortamiento según el segmento (rizomelia, mesomelia, acromelia, macromelia), evaluar la calidad ósea (mineralización, fracturas, deformidades, ausencia de huesos) evaluarse el cráneo fetal, su biometría y su osificación y morfología, la circunferencia abdominal, la mandíbula, la clavícula, la escápula, la circunferencia torácica. La ratio fémur/pie, que se mantiene en torno a 1 durante toda la gestación. También debe evaluarse el perfil (abombamiento glabelar, puente nasal plano, micrognatia), la cara y las orejas, los cuerpos vertebrales (platispondilia, mineralización, hemivértebras…), las manos y pies (braquidactilia, anomalías posicionales) y cantidad de líquido amniótico6,7.
Debe evaluarse si es una displasia letal o no letal. La letalidad ocurre en las displasias esqueléticas como resultado una circunferencia torácica pequeña que produce hipoplasia pulmonar. Desde el punto de vista ecográfico la ratio tórax/circunferencia abdominal <0,6 y la ratio fémur/circunferencia abdominal menor a 0,16 son altamente sugestivas de letalidad6–8.
El diagnóstico genético es el gold standard para el diagnóstico de las displasias esqueléticas7,9. Existe una página Web de libre acceso que puede orientarnos en cuanto al diagnóstico genético prenatal según los hallazgos o la sospecha diagnóstica, que es: https://www.genetests.org/
Desde el punto de vista morfológico el diagnóstico correcto oscila entre un 60-70% en los últimos 15 años, y variable según el tipo de displasia estudiada, siendo de un 80-90% para las anomalías asociadas a la alteración de la mineralización10.
El diagnóstico diferencial se debe realizar con acondrogénesis, osteogénesis imperfecta tipo ii, la hipofosfatasia tipo i y la displasia tanatofórica, que representan las displasias esqueléticas letales más frecuentes.
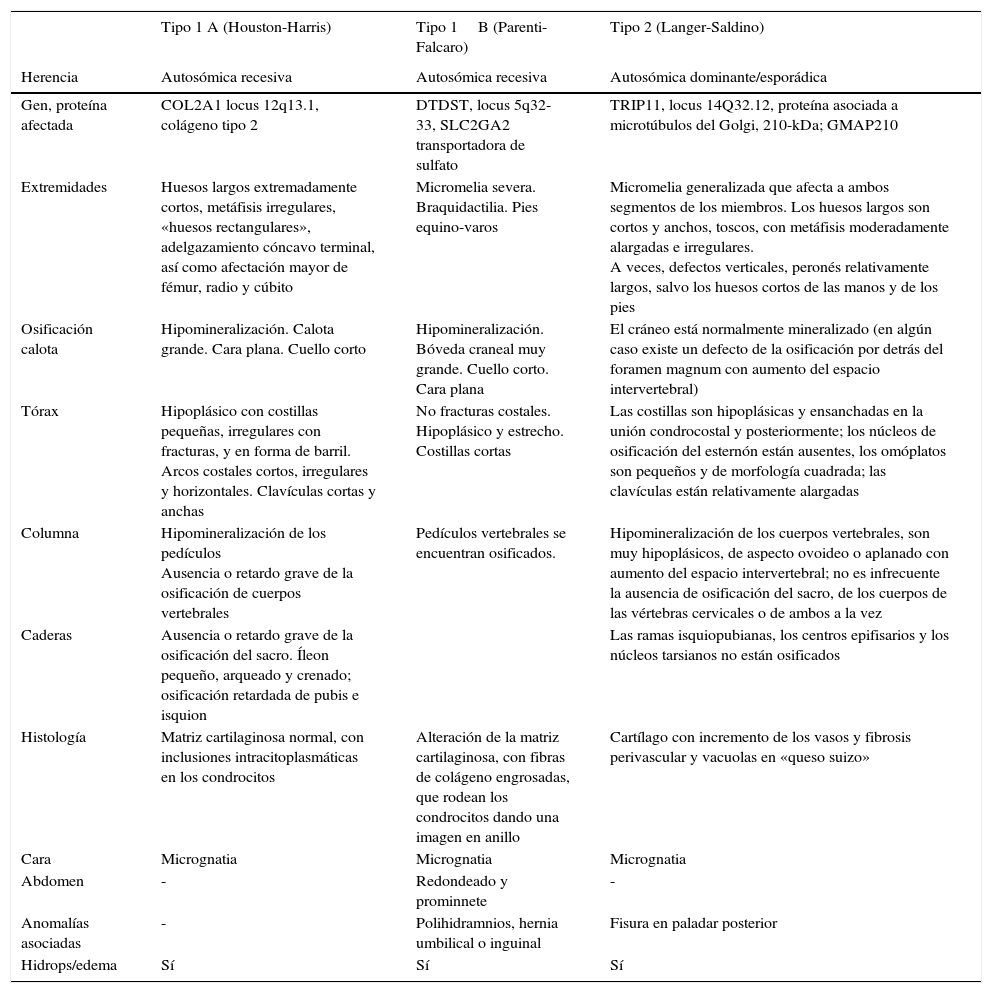
Las acondrogénesis son displasias esqueléticas letales que cursan con trastornos severos en el desarrollo del hueso y el cartílago11. La incidencia de la acondrogénesis es de 1/40.000 recién nacidos vivos5. Se divide en 2 tipos según los hallazgos clínicos y radiológicos. El tipo i o tipo Faccaro-Houston-Harris, el tipo ii o de Langer-Saldino. La tipo i se divide en 2 subtipos, la IA y la IB según criterios histológicos12. Las características clínico-radiológicas se describen en la tabla 1.
Características diferenciales clínico-radiológicas de los tipos de acondrogénesis
| Tipo 1 A (Houston-Harris) | Tipo 1B (Parenti-Falcaro) | Tipo 2 (Langer-Saldino) | |
|---|---|---|---|
| Herencia | Autosómica recesiva | Autosómica recesiva | Autosómica dominante/esporádica |
| Gen, proteína afectada | COL2A1 locus 12q13.1, colágeno tipo 2 | DTDST, locus 5q32-33, SLC2GA2 transportadora de sulfato | TRIP11, locus 14Q32.12, proteína asociada a microtúbulos del Golgi, 210-kDa; GMAP210 |
| Extremidades | Huesos largos extremadamente cortos, metáfisis irregulares, «huesos rectangulares», adelgazamiento cóncavo terminal, así como afectación mayor de fémur, radio y cúbito | Micromelia severa. Braquidactilia. Pies equino-varos | Micromelia generalizada que afecta a ambos segmentos de los miembros. Los huesos largos son cortos y anchos, toscos, con metáfisis moderadamente alargadas e irregulares. A veces, defectos verticales, peronés relativamente largos, salvo los huesos cortos de las manos y de los pies |
| Osificación calota | Hipomineralización. Calota grande. Cara plana. Cuello corto | Hipomineralización. Bóveda craneal muy grande. Cuello corto. Cara plana | El cráneo está normalmente mineralizado (en algún caso existe un defecto de la osificación por detrás del foramen magnum con aumento del espacio intervertebral) |
| Tórax | Hipoplásico con costillas pequeñas, irregulares con fracturas, y en forma de barril. Arcos costales cortos, irregulares y horizontales. Clavículas cortas y anchas | No fracturas costales. Hipoplásico y estrecho. Costillas cortas | Las costillas son hipoplásicas y ensanchadas en la unión condrocostal y posteriormente; los núcleos de osificación del esternón están ausentes, los omóplatos son pequeños y de morfología cuadrada; las clavículas están relativamente alargadas |
| Columna | Hipomineralización de los pedículos Ausencia o retardo grave de la osificación de cuerpos vertebrales | Pedículos vertebrales se encuentran osificados. | Hipomineralización de los cuerpos vertebrales, son muy hipoplásicos, de aspecto ovoideo o aplanado con aumento del espacio intervertebral; no es infrecuente la ausencia de osificación del sacro, de los cuerpos de las vértebras cervicales o de ambos a la vez |
| Caderas | Ausencia o retardo grave de la osificación del sacro. Íleon pequeño, arqueado y crenado; osificación retardada de pubis e isquion | Las ramas isquiopubianas, los centros epifisarios y los núcleos tarsianos no están osificados | |
| Histología | Matriz cartilaginosa normal, con inclusiones intracitoplasmáticas en los condrocitos | Alteración de la matriz cartilaginosa, con fibras de colágeno engrosadas, que rodean los condrocitos dando una imagen en anillo | Cartílago con incremento de los vasos y fibrosis perivascular y vacuolas en «queso suizo» |
| Cara | Micrognatia | Micrognatia | Micrognatia |
| Abdomen | - | Redondeado y prominnete | - |
| Anomalías asociadas | - | Polihidramnios, hernia umbilical o inguinal | Fisura en paladar posterior |
| Hidrops/edema | Sí | Sí | Sí |
La osteogénesis imperfecta tipo ii es de las displasias esqueléticas letales más frecuentes, junto con la displasia tanatofórica; su incidencia es de 1-2/100.000 y presenta una herencia autosómica dominante o recesiva, con defecto en el gen que codifica las proteínas alfa-1 procolágeno (COL1A), alfa-2 procolágeno (COLA2) localizadas en los cromosomas 7 y 175. Cursa con hipomineralización de la calota fetal, acortamiento de huesos largos por las múltiples fracturas asociadas, fracturas costales múltiples y tórax en forma de campana con hipoplasia torácica.
La hipofosfatasia tipo i es una enfermedad rara, con incidencia de 1/100.00012, autosómica recesiva, que presenta hipomineralización ósea, excepto en las clavículas, micromelia severa e hipoplasia torácica. Esto es causado por una baja o nula actividad de la fosfatasa alcalina inespecífica de tejido (TNSALP), causada por la mutación del gen ALPt localizado en el locus 1p36. 1-p345,6.
La displasia tanatofórica es otra entidad que debemos tener en cuenta. Tiene una incidencia de 1/100.000, y está causada por mutaciones del gen del receptor 3 del factor de crecimiento fibroblástico. Presenta macromelia severa, pero las extremidades son más largas que en la acondrogénesis, el tórax es hipoplásico, con cráneo en forma de trébol (en la tipoii) grande, con osificación adecuada y con frente prominente. Se diferencia del resto en que la mineralización no está afectada.
Es importante realizar un estudio multidisciplinar, genético, anatomopatológico, clínico y radiológico para llegar al diagnóstico y realizar asesoramiento. La identificación genética puede realizarse en 2/3 de los casos13. En nuestro caso el diagnóstico clínico y radiológico fue suficiente para establecer el pronóstico pero, lamentablemente, no se pudo completar el estudio del tipo exacto de acondrogénesis, para poder realizar asesoramiento apropiado a estas gestantes para futuras gestaciones. Por ello la importancia del conocimiento que debemos tener sobre el diagnóstico prenatal de estas enfermedades para evitar que esto pueda ocurrir.
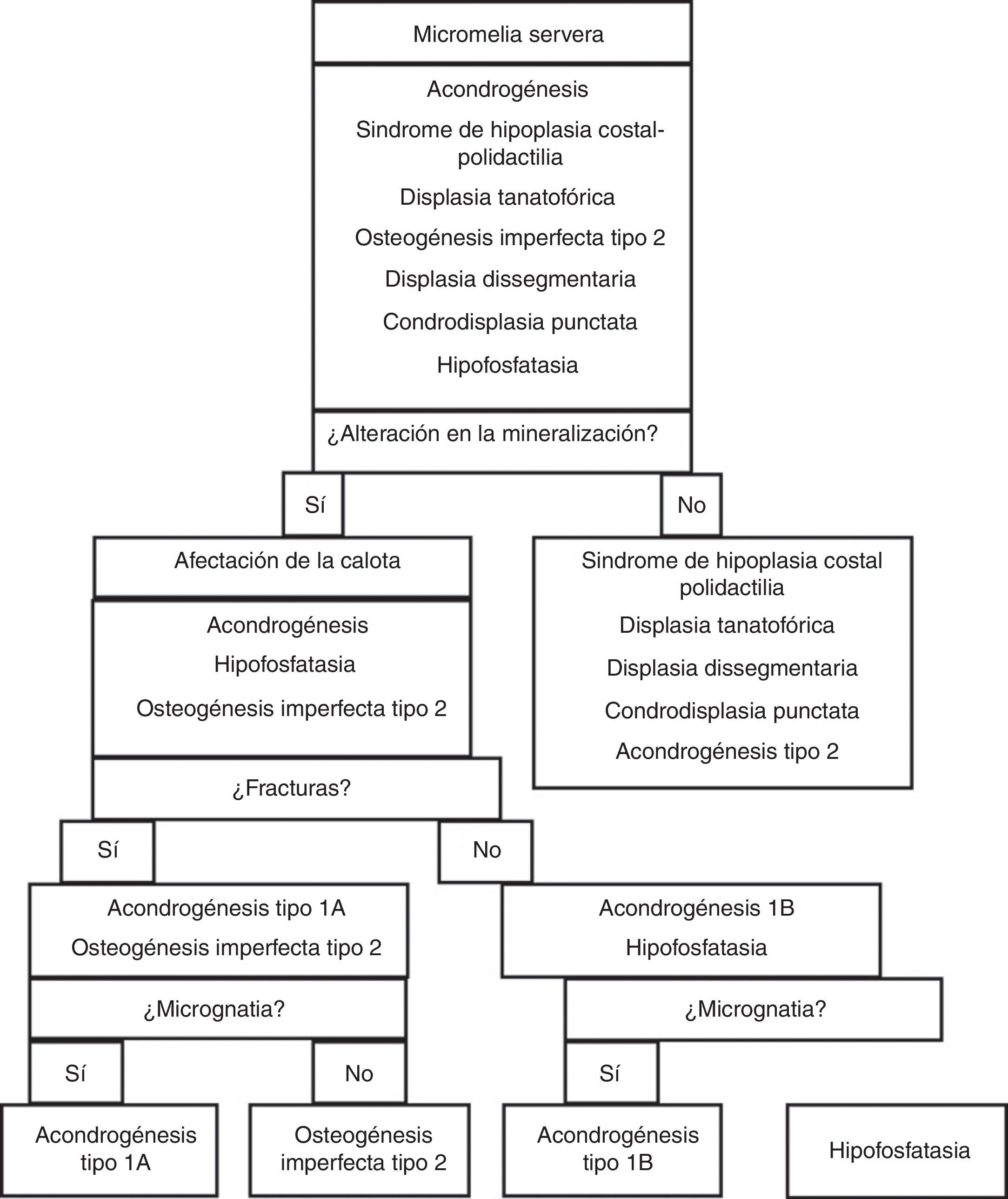
Se ha elaborado un algoritmo diagnóstico que nos permite orientar ante qué tipo de displasia con alteración de la mineralización nos encontramos (fig. 11).

Se ha valorado la utilidad de la ecografía 3D en el estudio de la cara fetal, la valoración del contorno nasal, las suturas, la micrognatia, la valoración de las manos y los pies y da mayor información que la ecografía 2D en estos casos8. En nuestros casos los hallazgos ecográficos obtenidos mediante la ecografía 3D nos fueron de gran ayuda a la hora de establecer el diagnóstico más probable.
Conflicto de interesesLos autores declaran que no presentan conflictos de intereses.
