



La hipertensión arterial pulmonar (HAP; grupoi de la clasificación de Dana Point)1 es una variante de hipertensión pulmonar (HP) caracterizada por reducción del lecho arteriolar pulmonar (hipertrofia de la media, proliferación de la íntima, fibrosis, inflamación perivascular y lesiones trombóticas) que conduce a incremento de la resistencia vascular pulmonar (RVP), deterioro de la función del ventrículo derecho y muerte. Hemodinámicamente se caracteriza por presión arterial pulmonar media (PAPm)>25mmHg, presión capilar pulmonar (PCP) normal (a diferencia del grupoii) y ausencia de enfermedad pulmonar (grupoiii) y de patología tromboembólica (grupoiv). La HAP puede ser idiopática (HAPI) o asociada a procesos como enfermedad del tejido conjuntivo (HAP-ETC), cardiopatías congénitas, infección por VIH, hipertensión portal, exposición a anorexígenos o toxinas, e HP persistente del recién nacido. La clasificación de Dana Point (2008) añadió la HAP asociada a esquistosomiasis (causa conocida más frecuente de HAP en el mundo) y a las anemias hemolíticas, ausentes de este grupo en la clasificación previa de Venecia. Por último, la HAP hereditaria (HAPH) se relaciona con mutaciones genéticas del receptor tipoii de la proteína morfogenética ósea2 (BMPR2), de la cinasa-1 análoga al receptor de activina (ALK1) o de otros genes desconocidos (familias con alta incidencia de HAP sin relación con dichas alteraciones). Existen diferencias en los mecanismos fisiopatológicos, el pronóstico y la respuesta al tratamiento entre las variantes de HAP. En los últimos años se ha avanzado en los métodos diagnósticos y el conocimiento de la genética, de la fisiopatología y de los mecanismos de acción de los medicamentos que son cruciales para mejorar el pronóstico de los pacientes.
Avances en la fisiopatologíaLas mutaciones citadas se relacionan con un número limitado de pacientes: de receptor de endoglina y ALK1 en casos de HAP asociada a enfermedad de Rendu-Osler y de BMPR2 en el 80% del grupo HAPH (que representa solo el 2,7% del total de HAP [estudio REVEAL]) y en el 10-20% de casos de HAPI. Recientemente se han descrito modificaciones para otros genes, como los que codifican el transient receptor potential channel 6 (TRPC6) o el trasportador de los canales de potasio Kv1.5 (KCNA5). La serotonina (5HT) estimula la proliferación de músculo liso en modelos experimentales. La mutación del gen de su transportador (5-HTT), frecuente en pacientes con HAPI, incrementa su efecto en células musculares vasculares. Existen indicios de relación entre polimorfismos de otros genes con HAP asociada a hipertensión portal o a esclerosis sistémica (ES); concretamente, para esta última, con el intrón-7 del gen de endoglina, con las variante rs10744676 del gen KCNA5, rs344781 del gen UPAR9 y con polimorfismos de IL-23. Estas modificaciones confieren, además de susceptibilidad, características especiales: las de BMPR2 se relacionan con menor edad de inicio y en el fallecimiento, mayor gravedad hemodinámica, menor respuesta a la prueba vasodilatadora y peores resultados con tratamiento; las de las de TRPC6 incrementan la activación del factor inflamatorio de transcripción nuclear, NF-B6, mientras que las de KCNA5 se asocian a HAP por consumo de fenfluramina. Se han detectado modificaciones epigenéticas (no modifican la estructura del ADN pero modulan su función sobre crecimiento, proliferación o apoptosis), tales como modificación de histonas, metilación del ADN o efectos de moléculas de microARN, que favorecen el desarrollo de HAP. Las modificaciones epigenéticas de superóxido dismutasa-2 (SOD2), óxido nítrico (NO) sintetasa y elementos de la cadena de BMPR2 son ejemplo de todo ello. Nuevas investigaciones establecen la posibilidad de actuar, a nivel postraslacional, sobre la deficiente expresión de BMPR2. Spiekerkoetter et al. comprobaron que el tacrolimus mejora la expresión de moléculas p-Smad 1/5/8 e Id1 (dependientes de la activación de BMPR2) y la supervivencia de células endoteliales y previene la aparición de HP y de hipertrofia del ventrículo derecho en modelos experimentales. Efectos semejantes se han observado para la rapamicina. Actualmente un grupo europeo-estadounidense trabaja sobre una colección de especímenes de ADN de pacientes con HAP para investigar factores genéticos y epigenéticos asociados y respuesta a tratamientos2.
Avances en el diagnóstico de la hipertensión arterial pulmonarLa sintomatología inicial de la HAP, aparte de inespecífica (disnea, insuficiencia cardiaca derecha), es silente durante mucho tiempo. Habitualmente el diagnóstico llega muy tardíamente (en los registros francés [FNPH], estadounidense [REVEAL] y español [REHAP] los pacientes estaban en clase funcional [CF] III-IV en el 75, el 73 y el 70% en ese momento).
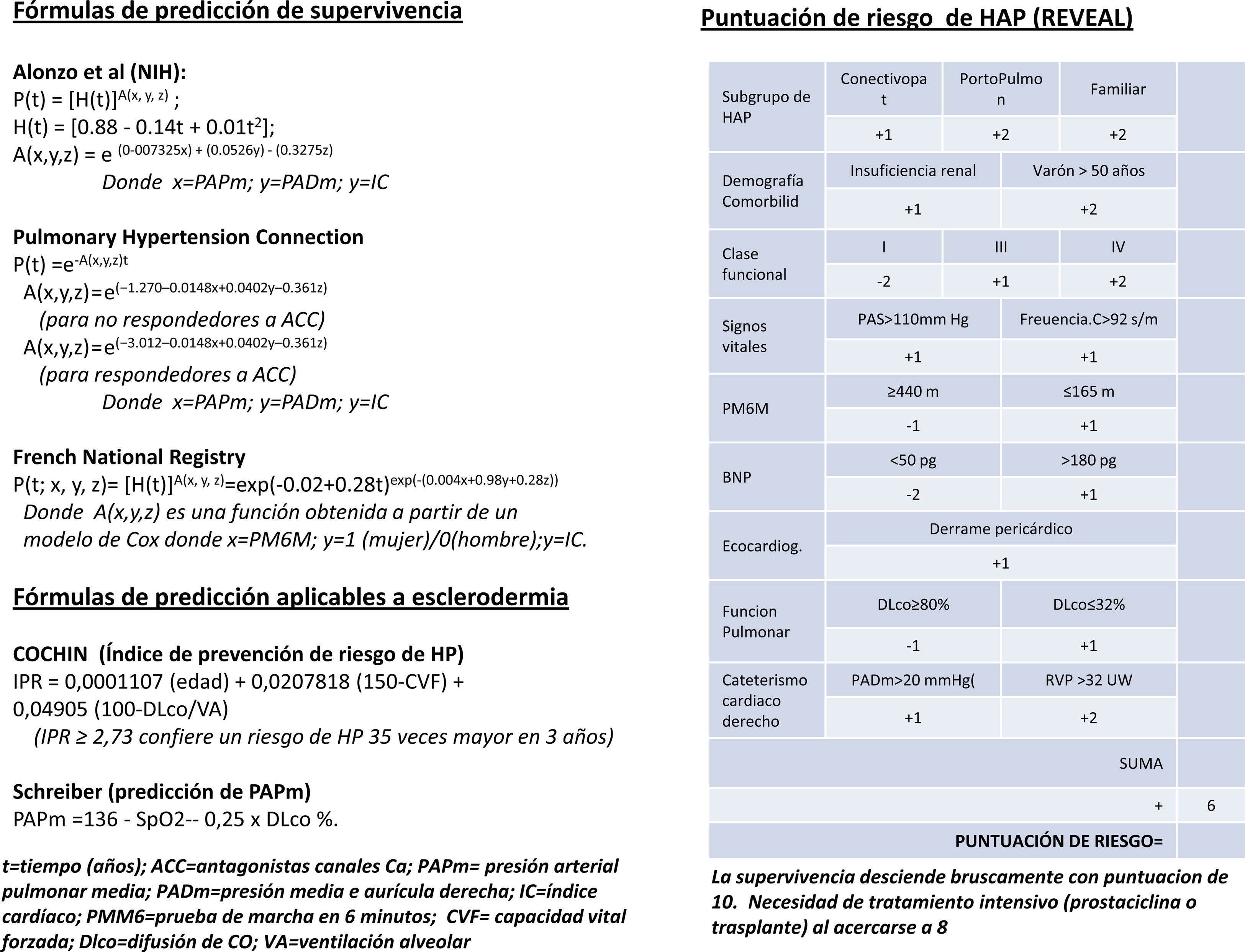
Mientras que para la HAPI es muy difícil mejorar el retraso diagnóstico, en pacientes pertenecientes a grupos de riesgo, como HAP-ETC (25% del total de HAP), los programas de rastreo mediante ecocardiografía transtorácica (ECGTT) permiten un diagnóstico precoz y, por lo tanto, un tratamiento más temprano que influyen favorablemente en la supervivencia. Un paso más es el intento de diagnóstico (de presunción) de HAP en pacientes con ETC con disnea no explicada y con PAPs normal en reposo. El ECGTT de esfuerzo (ECG-Ef) permite detectar estadios subclínicos de HAP. Aunque las guías clínicas actuales han suprimido el criterio de PAPm en esfuerzo (PAPm-Ef)>30mm Hg como diagnóstico de HP, el empleo de ECG-Ef como método incruento de cribado ha resultado de gran utilidad (Steeen et al., 2008; Callejas et al., 2008). El perfeccionamiento de la ECGTT ha hecho posible diferenciar, con bastante aproximación, entre HAP e HP secundaria a disfunción diastólica del ventrículo izquierdo, ambas asociadas a ES (la ETC más frecuentemente asociada a HP), ya que permite valorar, además de la PAPm (mediante la fórmula de Chemla: PAPm=PAPs0,6+2), la PCP (a partir del cociente E/e’), el GC (a partir de la integral velocidad/tiempo del tracto de salida del ventrículo izquierdo) y la RVP combinando los valores anteriores3. Todo ello analizado con más detalle en otro apartado de este número de la revista. Para la ES, se han desarrollado 2fórmulas para calcular el riesgo de desarrollar HP (la del Hospital Cochin [Meune et al., 2011] y la de Schreiber et al., 2011) a partir de valores funcionales respiratorios (figura 1).
Avances en la valoración de actividad, la eficacia del tratamiento y la predicción de supervivencia
Contamos con diferentes índices de gravedad y de eficacia del tratamiento en HAP: prueba de la marcha de 6min, CF-OMS, escala de disnea de Borg, parámetros ecocardiográficos (índice de TEI, TAPSE) o datos hemodinámicos obtenidos mediante cateterismo cardiaco derecho. En una revisión reciente4 se analiza el valor predictivo de diferentes marcadores biológicos, algunos con valor establecido, como péptidos natriuréticos y troponina-T, y otros en fase de investigación, como proteína transportadora de ácidos grasos específica del miocardio, endotelinas, dímeros D, factor Von Willebrand, dimetil-arginina asimétrica o ligando10 de quimiocinaCXC (CXCL10). La elevación de determinadas citocinas (TGF-15, IL-6, IL-8, IL-10 e IL-12p70), las modificaciones de angiopoyetina-2 y de la matriz-metaloproteinasa-9 o la presencia de niveles elevados de la micropartícula endotelial CD62e+ se relacionan con el grado de inflamación. Se han comprobado diferencias cuantitativas en la expresión de proteínas del pulmón de pacientes con HAP en comparación con controles, bien por exceso (periostina, CLIC-1, CLIC-4), bien por defecto (haptoglobina, vinculina), lo que abre nuevas posibilidades en el conocimiento de la patogenia y nuevos marcadores de la HAP4.
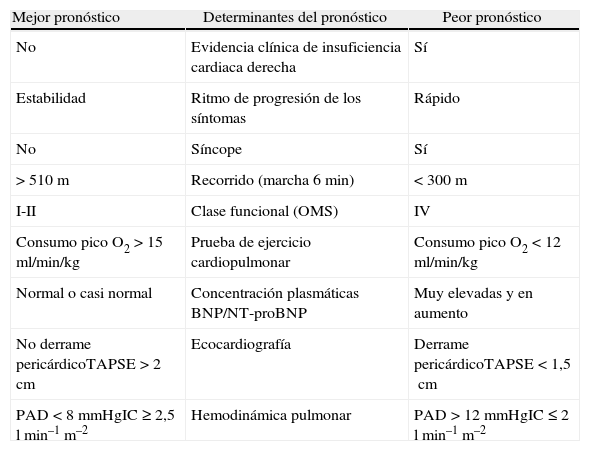
Se han utilizado diferentes fórmulas para la predicción de supervivencia. La de los NIH, de 1981, fue útil solo para pacientes no sometidos a tratamiento. En la actualidad, los datos de diferentes registros (REHAP, REVEAL, PHC y FNPH) demuestran un considerable incremento de la supervivencia coincidente con el advenimiento de los tratamientos específicos. A partir de los 2 últimos, se han desarrollado nuevas fórmulas (figura 1) para el cálculo de la supervivencia en el momento del diagnóstico (en pacientes en la era actual, en la que, nuevamente, existen tratamientos específicos). Además, a partir del registro REVEAL se ha desarrollado una herramienta para calcular el riesgo, al inicio del diagnóstico y seriadamente, que es de gran utilidad para adaptar el tratamiento (figura 1). La tabla 1 recoge los parámetros que permiten considerar a un paciente en situación estable y satisfactoria (columna de la izquierda), inestable e insatisfactoria (columna de la derecha) o estable pero insatisfactoria (situación mixta). Las 2 últimas indican la necesidad de intensificar el tratamiento.
Parámetros para considerar la situación de un paciente
| Mejor pronóstico | Determinantes del pronóstico | Peor pronóstico |
| No | Evidencia clínica de insuficiencia cardiaca derecha | Sí |
| Estabilidad | Ritmo de progresión de los síntomas | Rápido |
| No | Síncope | Sí |
| >510m | Recorrido (marcha 6min) | <300m |
| I-II | Clase funcional (OMS) | IV |
| Consumo pico O2>15ml/min/kg | Prueba de ejercicio cardiopulmonar | Consumo pico O2<12ml/min/kg |
| Normal o casi normal | Concentración plasmáticas BNP/NT-proBNP | Muy elevadas y en aumento |
| No derrame pericárdicoTAPSE>2cm | Ecocardiografía | Derrame pericárdicoTAPSE<1,5cm |
| PAD<8 mmHgIC≥2,5lmin–1m–2 | Hemodinámica pulmonar | PAD>12 mmHgIC≤2lmin–1m–2 |
De Barberá et al. Estándares asistenciales en hipertensión pulmonar. Rev Esp Cardiol. 2008;61:170–84.
La vías de actuación principales en el tratamiento de la HAP son la de los prostanoides, la de los antagonistas de receptores de endotelina (ARE) y la del NO.
La introducción del epoprostenol (EPP) intravenoso, análogo de la prostaciclina, supuso un avance radical en el tratamiento de la HAP. Otros prostanoides, como el treprostinil (TPT), subcutáneo, o el iloprost (IP), inhalado, permitieron obviar problemas inherentes a la administración intravenosa. No obstante, el EPP es el tratamiento de elección en CFIV. El selexipag, análogo oral de la prostaciclina y agonista oral del receptor de prostaglandinaI2. (IPr), es altamente selectivo, lo que puede reducir efectos adversos al no activar receptores como EP3, cuyo efecto es la vasoconstricción arterial pulmonar, que sí son activados por otros prostanoides. De eficacia comprobada en faseii, hay 2 ensayos clínicos controlados (ECC) en marcha.
Actualmente se utilizan 2ARE: el bosentán y el ambrisentán (tras la retirada del sixtasentán, por hepatotoxicidad). El macitentán, un nuevo ARE, se diferencia de los precedentes por su distribución tisular (mejor penetración por lipofilia), por su mayor capacidad de unión al receptor y por presentar menores interacciones medicamentosas, lo que se traduce en dosis reducidas y mejor tolerancia. Hay un ECC en faseiii en curso.
Los inhibidores de fosfodiesterasa-5 (IPD5) incrementan la concentración de guanosinmonofosfato cíclico (GMPc; dependiente este de la producción de NO, estimulante a su vez de guanosinciclasa [sGC] que activa su síntesis) bloqueando su trasformación en GMP. El sildenafilo y el tadalafilo son sus principales representantes. El riociguat estimula también la síntesis de sGC dependiente de NO pero, además, activa directamente dicha síntesis, lo que permite, a diferencia de los IPD5, incrementar el GMPc en situaciones de subproducción de NO.
Diferentes guías establecen la posibilidad de tratamiento combinado en casos de resistencia a monoterapia, pero últimamente se plantean estrategias más intensas. Algunos ensayos clínicos en marcha (AMBITION) valoran el resultado de tratamiento inicial combinado versus monoterapia. En el estudio EARLY se comprobó una evolución más favorable cuando se inicia el tratamiento en pacientes poco evolucionados (CFII). Kovacs, en HAP-ES, recomienda introducirlo en fases aún más precoces: HAP borderline (PAPm normal en reposo, con PAPm-Ef>30mmHg). Comprueban una evolución más favorable que en controles. Para el otro extremo del espectro de la enfermedad, HP en fases avanzadas en formas de especial mal pronóstico (ES, HAPH), se aconseja hacer «algo más» que lo que recomiendan las guías. Kemp sugiere que, en pacientes en CFIII-IV, cuya situación basal es especialmente grave, iniciar de novo tratamiento combinado, con EPP+bosentán, es más eficaz que la monoterapia con EPP. En 23pacientes con mal pronóstico basal (16 en CFIII y 7 en CFIV), tratados con dicha combinación, su evolución a los 4meses fue más favorable que la de controles en monoterapia. Sitbon y Simonneau observan que pacientes especialmente graves (incluso en CFIII pero con criterios de gravedad) deben someterse, en primera línea, a tratamientos combinados (doble o triple terapia incluyendo EPP).
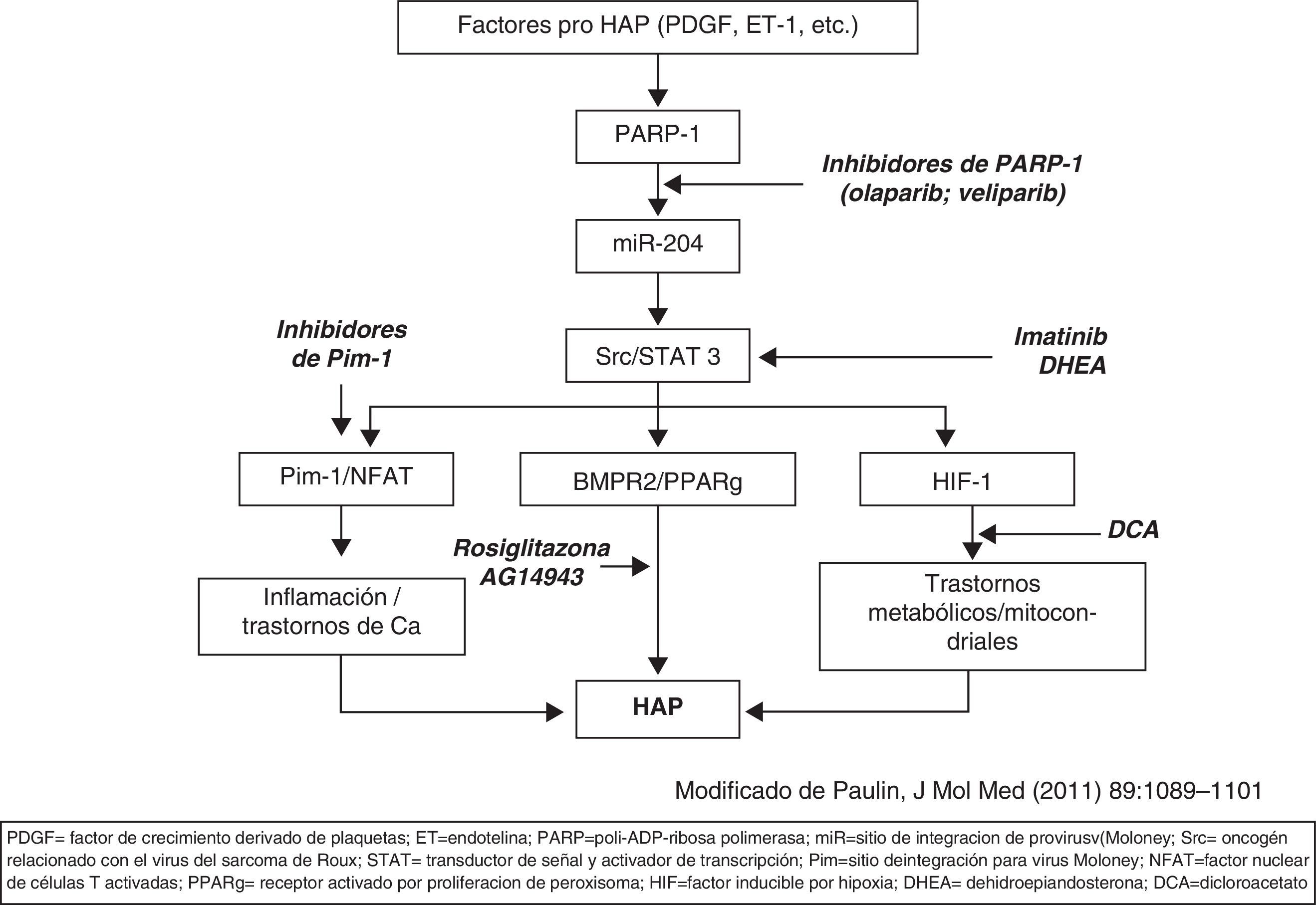
Se están desarrollando nuevas vías de tratamiento (la mayoría en fase experimental) que actúan paralelamente sobre procesos implicados en el desarrollo de HAP distintos de las vías mencionadas. El imatinib, inhibidor de receptores de PDGF y cKit, es un agente antiproliferativo que se ha comprobado eficaz en ensayos clínicos en faseii; está en marcha otro ensayo clínico en faseiii. Diferentes factores con acción pro-HAP (endotelina, angiotensina, PDGF, citoquinas, etc.) actúan sobre una cadena de moléculas constituidas por PARP-1, miR-2001 y Src/STAT3 que modulan la actividad de: a)Pim-1/NFAT; b)BMPR2/PPAR, y c)HIF1. El efecto de estas 3 acciones potencia el desarrollo de HAP. En la figura 2 (modificada de Bonnet y, a su vez, de Paulin et al.) se esquematizan posibles nuevas dianas en el tratamiento de la HAP.

La HAP representa el 4% de causas de trasplante pulmonar o cardiopulmonar (indicado en pacientes refractarios). La supervivencia a los 5años es del 47%. La progresiva mejoría en la supervivencia lograda tanto con tratamiento médico como con el avance de las técnicas quirúrgicas obliga a una cuidadosa valoración para pronunciarse por una u otra variante de tratamiento.
Conflicto de interesesLos autores no tienen conflictos de intereses en relación con este artículo.

