












Pagina nueva 1
1. Summary of the clinical history (A-12-26)
A 4-year-old male patient was referred to a second level of care hospital due to increased size of submandibular, axillary and inguinal lymph nodes of 2 months duration accompanied by groin pain and fever up to 39 °C.
1.1. Nonpathological history
The patient was originally from and a resident of Zumpango, State of Mexico, with a low socioeconomic status.
1.2. Perinatal and past medical history
The patient was the product of a first pregnancy. The mother reported regular prenatal visits and early labor at 24 weeks gestation, which was managed with rest. The patient was born at term abdominally due to the cord being wrapped around the neck. He weighed 3,300 g and measured 51 cm. The infant was discharged without complications.
1.3. Current illness
November 7-23, 2011. At 4 years 4 months of age the patient was referred to Hospital Infantil de México Federico Gómez (HIMFG) due to enlarged lymph nodes. The initial encounter is for an outpatient consultation (OC) with general surgery with left cervical node biopsy. A lymph node conglomeration was found compatible with Castleman’s disease (CD) (hyalinovascular type) by immunohistochemical staining (CD10, CD20, CD3 and BCL-6, positive; LMP-1 and CD30, negative), bone marrow aspirate was negative for neoplastic infiltration.
December 26-31, 2011. Bilateral radical cervical lymphadenectomy was performed with histopathological result of hyalinovascular CD.
February 16-March 26, 2012. The patient was admitted to the Emergency Department due to illness of 15 days evolution with dysphagia of moderate intensity, painful whitish plaques in the mouth that easily came off as well as productive cough, cyanosis and dyspnea of 24-h evolution, accompanied by fever of 16 h. On physical examination, desaturation and crepitant rales were found in the right hemithorax, as well as bilateral parahilar infiltrate on chest x-ray. The patient was treated with cefotaxime, clindamycin and fluconazole. Due to respiratory and hemodynamic deterioration he required management in the Pediatric Intensive Care Unit (PICU) with boluses of crystalloids, norepinephrine, levosimendan and mechanical ventilation for 3 days, with noted improvement. He subsequently presented nosocomial sepsis and persistence of the oral cavity lesions for which he was managed with cefepime (10 days), amphotericin B (10 days) and acyclovir (7 days). He was admitted to the Department of Internal Medicine for continuation of management and studies. Upon dermatology examination, the patient was found to have generalized dermatosis with erythematous scaly plaques, papules and bloody, itchy scabs. Cetirizine and white cream was ordered. Computed tomography (CT) was done on March 6 where bilateral reticulonodular infiltrate was seen at the pulmonary bases as well as hepatomegaly, multiple lymphadenopathies at the bilateral axilla, retroperitoneum, obturator chain and the bilateral inguinal region up to 2.1 cm in diameter cross-sectionally. The patient was evaluated by the Oncology Department because of increase in size and number of the lymph nodes, hepatomegaly, fever in the evening and night, weight loss and diaphoresis.
April 3, 2012. In the OC of the Oncology Department, the patient was started on prednisone at a dose of 2 mg/kg/day.
April 24, 2012. During the follow-up visit, the patient persisted with cervical, inguinal and axillary lymphadenopathies. CBC showed leukocytes (60,300) with 84% neutrophils. He was continued on prednisone.
May 14-18, 2012. In the OC of the Oncology Department, an important reduction in lymphadenopathy was noted with acute tonsillitis and mouth lesions suggestive of oral candidiasis. He was treated with azithromycin and fluconazole. Control CT documented consolidation in the posterior segment of the right upper lobe, multiple lymph nodes in retroperitoneum (1.4 cm × 1.8 cm) at both iliacs (2.4 × 2 cm and 2.9 × 1.9 cm) and inguinofemoral region (2.1 × 1.8 cm) with discreet decrease in size compared to the study carried out in March.
May 19, 2012. The patient was admitted to the Emergency Department due to epistaxis of 6 days evolution, nonprogressive cough of 4 days evolution with insidious onset predominantly in the mornings with greenish expectoration and in the last hours with respiratory difficulty. The patient had fever of 3 days evolution of up to 40 °C with diaphoresis, piloerection and chills. He also had painful oral lesions of 4 days evolution. On physical examination the following vital signs were noted: cardiac rate 179/min, respiratory rate 26/min, blood pressure 117/61 mmHg, temperature 39.9 °C, saturation 85%. Cushingoid facies, oral cavity with whit-ish, indurated painful, nonbleeding, nondetachable plaque in the right cheek and lower gums, oropharynx with posterior hyaline discharge, pulmonary fields with right basal rales, and soft, painful, mobile cervical, axillary and bilateral inguinal lymph nodes. Hepatomegaly was noted at 4-3-3 cm below the costal margin. Laboratory and imaging studies reported: leukocytosis 27,600, neutrophils 85%, bands 8%, hemoglobin 10.5 g/dl and hematocrit 31%. Chest x-ray demonstrated mixed infiltrate of the right basal and middle lobes.
The patient was managed with fasting, saline solution (1,800 ml/m2 SC/day, 2:1), potassium (30 mEq/m2/day), prednisone (75 mg/m2 SC/day), amoxicillin/sulbactam (80 mg/kg/day) and fluconazole (3 mg/kg/day). Nebulizations of salbutamol were given and due to respiratory deterioration with bronchospasm non-invasive mechanical ventilation was started. Antibiotic scheme was changed to cefotaxime and dicloxacillin. The patient was seen for consultation by ophthalmology for contact keratoconjunctivitis. Management with topical tobramycin/dexamethasone was started.
May 25, 2012. Fever continued. Chest x-ray demonstrated cottony infiltrates, and slides of the oral cavity lesion were positive for blastoconidias. Antibiotics were changed to cefepime, amikacin and amphotericin B.
May 27, 2012. The patient was referred for consultation by the Dermatology Department for dermatosis of the trunk and perianal region with a circumscribed area of denuded and necrotic skin of 7 days evolution. The condition affected the face at the level of the mouth and oral mucosa with multiple ulcerated plaques with a whitish bed at the base of the tongue, cheek and gums. Treatment was begun with bicarbonated water every 6 h and topical cicalfate.
May 29-June 4, 2012. The patient presented hemodynamic and respiratory deterioration due to persistent broncho spasm. He required intubation and initiation of norepinephrine, meropenem, vancomycin and amphotericin B lipid complex. Histopathological study of the bronchial aspirate was done where no fungi, bacteria or viral inclusions were seen. He was admitted to the PICU. Amines were removed after 24 h. CT of the chest noted “patches” of diffuse consolidation in the right and left hemithorax, air bronchogram, heterogeneous enhancement and diffuse alveolar infiltrate. Laboratory results were hemoglobin 7.7 g/ dl, hematocrit 23.6%, leukocytes 9,600, segments 78%, bands 2%, lymphocytes 17%, platelets 231,000. Galactomannan antigen and Mannan Candida antigen were negative. He had fresh blood coming from the endotracheal tube; 72 h later he had fever, hemorrhagic-necrotic lesions of the lips and multiple foci pneumonia. Trimethoprim-sulfamethoxazole was added to the management.
June 5-8, 2012. Fever persisted. There was fresh blood coming from the endotracheal tube. Oncology continued management exclusively with steroids. A new CT was done and demonstrated areas of bilateral consolidation seen in a prior study. Multiple axillary, mediastinal, common iliac chain and left internal lymphadenopathies were seen, the largest 2 cm × 2.4 cm. Lymph node conglomerates were demonstrated in the retroperitoneum and inguinal regions, as well as hepatosplenomegaly. The patient was febrile and with persistence of submandibular, axillary and inguinal adenopathy. No fungal structures were seen from bronchial aspirate. ELISA for HIV was nonreactive. Serology was as follows: Epstein-Barr virus: VCA IgM 14.6 (negative), VCA IgG 165 (positive), EA IgG 7.1 (negative), EBNA IgG 59.5 (positive). The patient was extubated on a programmed basis.
The Chest Surgery Department performed an endoscopy and reported the oral mucosa to be very friable with abundant whitish plaques at the cricopharynx and with necrosis of the esophageal mucosa in the upper third and middle. There were color changes in the lower third and bleeding friable mucosa. A biopsy was obtained. The patient was admitted intubated to the PICU for airway protection. Ophthalmology followed the case, noting blepharoedema, greenish membranes in the entire ocular surface with symblepharon formation and cloudy cornea, for which topical moxifloxacin and prednisone were started.
June 9-12, 2012. The patient experienced respiratory deterioration. He progressed to high-frequency oscillatory ventilation. Infusion with aminophylline, norepinephrine and milrinone was begun. Oxemia of 68%, PVC 9 mmH2O, venous reserve 75, Kirby 86. There was bleeding from the orotracheal cannula without improvement in ventilatory parameters. Ophthalmology suggested ruling out paraneoplastic pemphigus for presenting membrane forming acute follicular conjunctivitis. There was blepharoedema with abundant and thick membranes in the surface of the eye, which bled when removed. Laboratory results were hemoglobin 11 g/dl, hematocrit 26%, leukocytes 19,500, segments 89%, bands 3%, platelets 398,000, PT 12.9”, PTT 36.8”, INR 1.04, fibrinogen 495 mg/dl. Creatinine was 1.3 mg/dl, sodium 126 mEq/l, potassium 3.8 mEq/l, calcium 6.8 mg/dl, phosphorus 7.9 mg/dl, albumin 1.5 g/dl. The patient continued with hemodynamic deterioration, fresh bleeding from the nose, mouth and orotracheal tube, urinary catheter with blood clots, petechiae on the chest, generalized edema and oliguria. Chest x-ray demonstrated bilateral patchy radiopacity and data of active pulmonary hemorrhage. Norepinephrine was increased and a furosemide infusion was begun.
June 13, 2012. Adrenaline was initiated due to hemodynamic deterioration secondary to respiratory failure and profuse bleeding. The patient had anuria and continued to be hypotensive progressing to cardiorespiratory arrest. Advanced resuscitation maneuvers were carried out without response.
2. Imaging presentation
2.1. Coordinator (Dra. Martha Avilés Robles)
It is important to remember the approach of a patient with lymphadenopathies and the time at which a biopsy is indicated.
2.2. Imaging (Dra. Bertha Lilia Romero)
In the ultrasound of the cervical region carried out on February 23, 2012, there were multiple adenopathies identified on the right side of the lymph node chain. The Doppler effect showed a higher vascular flow and displacement of the adjacent vascular paths. On the left side there were lymph nodes >1 cm in size and similar to the right side showing a greater vascular flow in its center (Fig. 1). Simple chest x-ray of November 19 showed normal characteristics. Only a volume increase in both cervical regions in relation to lymph node growth was identified (figure not shown).
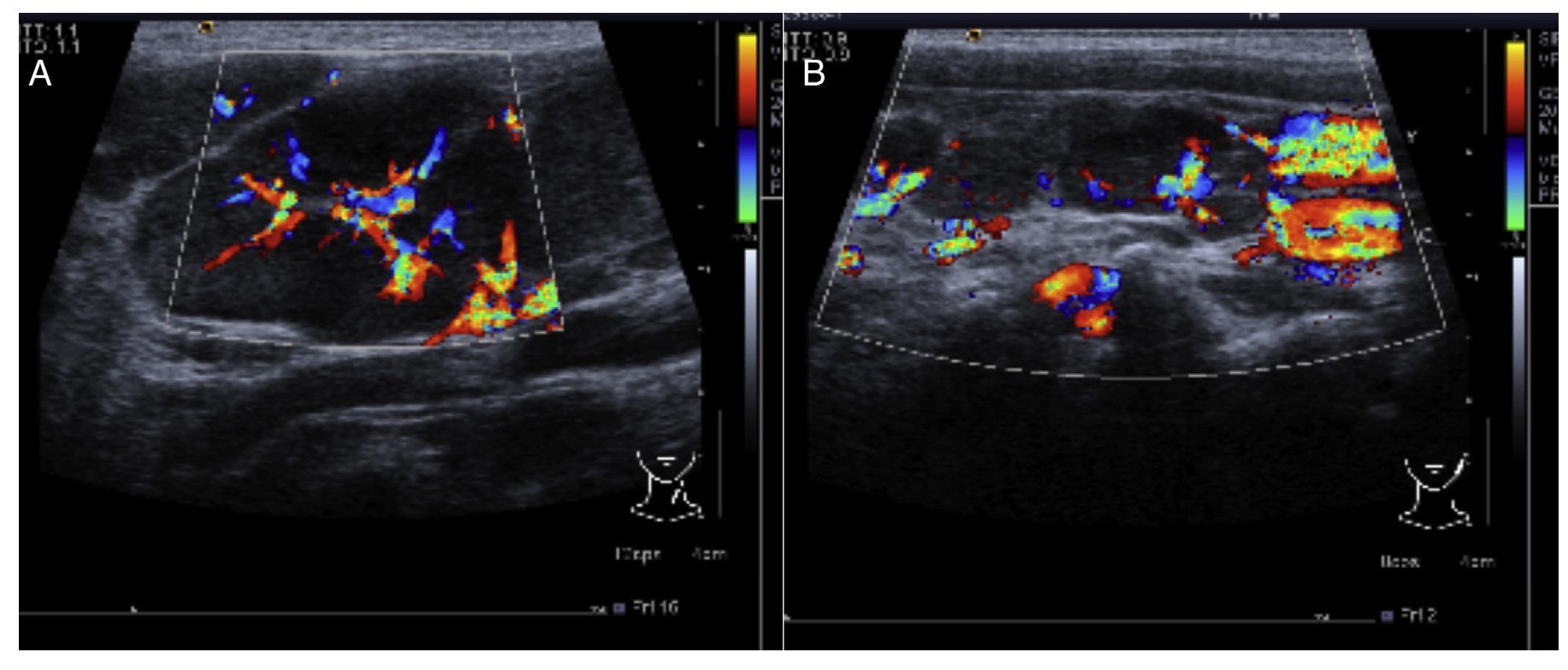
Figure 1 Ultrasound image in the cervical region bilaterally. Multiple adenopathies are observed at this level which, according to the Doppler effect, showed an increase of the flow within. (A) Left side. (B) Right side.
In another x-ray, diffuse radiopacity of right lung predominance was identified, which showed an air bronchogram in its interior, extending from the apical lobe to the base of the same lung, with probable zones in the parahilar region related with a pneumonic process. The cardiophrenic and costodiaphragmatic angles are free. The cannula was identified inside the trachea in good position. The remainder of the structures did not reveal any apparent changes (Fig. 2). In a follow-up study, a thoracoabdominopelvic projection was taken in which persistence of areas of consolidation previously mentioned are noted as well as air fluid levels (Fig. 3).
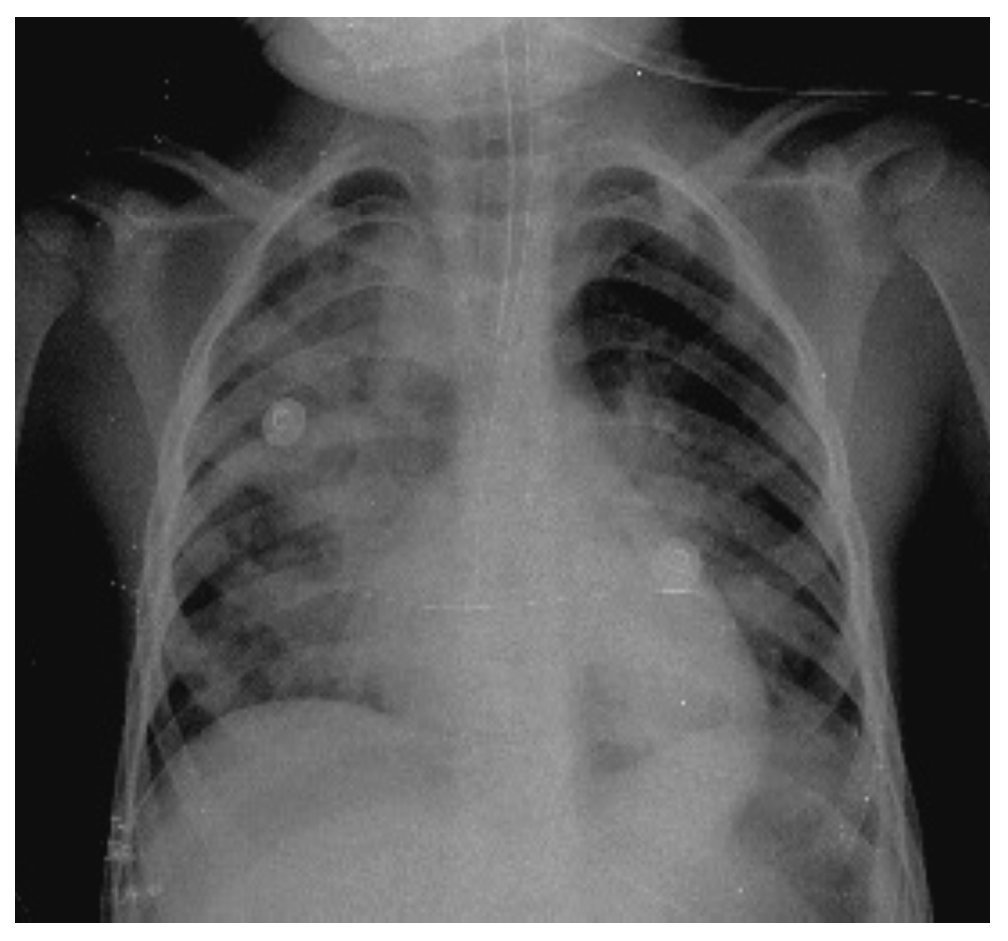
Figure 2 Chest study where aliveolar-type nfiltrate is identified in both pulmonary fields with right predominance.
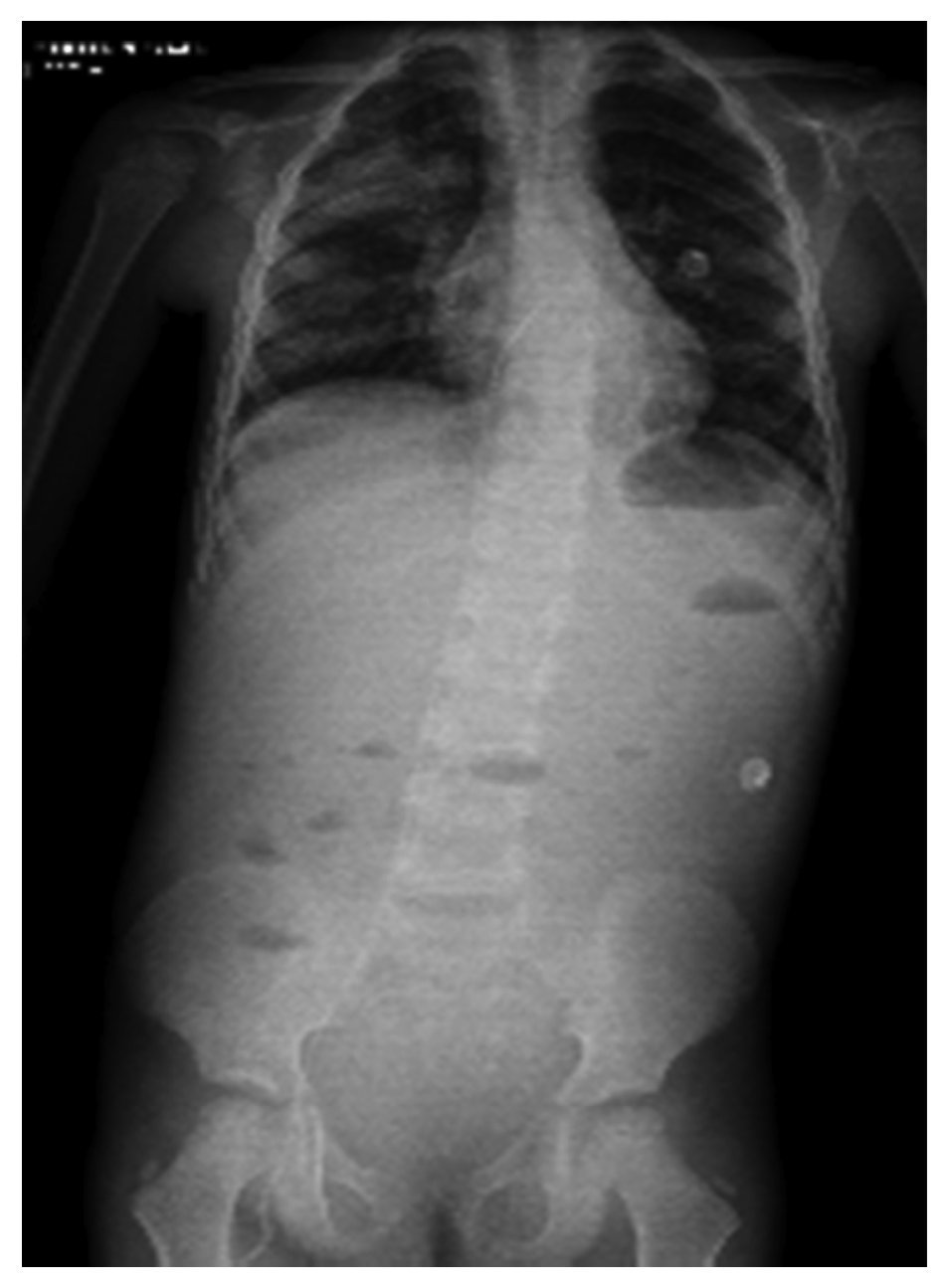
Figure 3 Thoracoabdominal projection with pneumonic foci in the right lung and air levels in the intestine with distal air.
3. Discussion
3.1. Emergency Department (Dr. Samuel Roberts Vega)
The subject is a pre-school male who was referred due to adenopathies and whose cervical lymph node biopsy was compatible with CD. Upon his last admission he was consulted for fever, cough, respiratory difficulty and epistaxis. The following syndromatic diagnoses were made:
1. Respiratory failure syndrome based on the presence of respiratory difficulty, right basilar crepitant rales, nasal flaring and oxygen saturation of 85%.
2. Systemic inflammatory response syndrome on the basis of tachycardia with 179 beats/min, fever up to 40 °C and leukocytosis of 27,600/mm3.
3. Immunodeficiency syndrome based on the medical history including multiple bacterial and fungal infections, weight loss and dermatosis.
4. Cushing syndrome according to the history of corticosteroid administration and characteristic facies.
5. Myeloproliferative syndrome according to the presence of multiple lymphadenopathies with lymph node conglomerates in the retroperitoneum and hepatomegaly.
After supporting the syndromatic diagnoses, the following nosologic diagnoses were made:
1. CD of mixed type or plasma cells with myeloproliferative syndrome initially with predominance of cervical lymphadenopathy and later on multiples areas, as well as according to the lymph node biopsy report.
2. Severe community-acquired pneumonia based on signs of respiratory failure, desaturation, crepitant rales, mixed basilar infiltrate and right cardiac arrest, as well as leukocytosis. According to the guidelines of the IDSA for community-acquired pneumonia in the pediatric age, the patient met the following severity criteria: hypoxemia, increase in respiratory effort, multilobar infiltrate, presence of co-morbidity and need for mechanical ventilation.
3. Sepsis based on the systemic inflammatory response syndrome and the presence of an infection, in this case pulmonary.
4. Septic shock based on the presence of sepsis and hemodynamic instability. The latter was supported by tachycardia, weak pulses, 3-sec capillary refill and the need for administration of vasoactive amines during the hospitalization course.
5. Pulmonary hemorrhage supported by radiological imaging compatibles with this disorder and obvious bleeding via the orotracheal cannula.
6. Acute respiratory distress syndrome. According to the Berlin criteria (2011), the patient presented deterioration of the respiratory symptoms, bilateral opacities and Kirby index of 178.
7. Multiple organ failure was determined on the basis of renal damage, respiratory failure and hemodynamic instability, i.e., state of shock.
The diagnoses and relevant data in the evolution of the patient are discussed below. In first place, diagnosis of CD was agreed although it is a disorder rarely present in children. This disease, according to the histopathological findings from the lymph nodes involved, was classified as hyaline vascular type. Even though in this patient the biopsy reported a hyaline vascular pattern, this type of disease usually manifests itself as unicentric and patients are asymptomatic. In this case, disease evolution corresponded more to the variety of plasma cells whose presentation is multicentric and is accompanied by systemic symptoms such as anemia, weight loss, fever, hepatomegaly, hemoptysis, skin lesions and, in a more severe presentation, can be fatal if not treated.1-5 Due to the appearance of the atypical presentation, initiation of steroids in the outpatient consultation appeared adequate although subsequently it caused a Cushingoid appearance as well described as part of the treatment in the multicentric presentation. Nevertheless, once the patient was hospitalized and progressing unfavorably, use of rituximab should have been considered, which is an anti-CD20 monoclonal antibody present in lymphoid cells and indicated as monotherapy or in combination with thalidomide.
Although the CD etiology is uncertain, there is an association between HIV and infection by type 8 human herpes virus and the latter with Kaposi sarcoma.2 Systemic fungal infections, cottony infiltrates, lesions in the oral cavity and esophagus which, although not able to be demonstrated via complementary tests, had their clinical effect to the degree of having fluconazole and amphotericin B administered to the patient. Oral and esophageal symptoms manifested could have been related to the probable immunocompromise and with the use multiple antibiotics in the OC.
The skin lesions described could be compatible with Kaposi sarcoma. There was also no skin biopsy taken that would confirm the diagnosis or PCR for type 8 human herpes virus. This entity could also have pulmonary manifestations such as dyspnea, wheezing and/or hemoptysis, which would be clinically compatible with the pulmonary hemorrhage the patient presented.
Paraneoplastic pemphigus should be included among the differential diagnoses of skin lesions. It is present in up to 10% of patients with CD. It is clinically characterized by a severe oral involvement consisting of stomatitis and polymorphic skin lesions mainly affecting the trunk, extremities, palms and soles. It can affect any mucosa of the organism, even developing severe pseudomembraneous colitis as was present in this patient. Paraneoplastic pemphigus is the only clinical type of pemphigus that is manifested in tissues not covered by stratified squamous epithelium, giving rise in ~30-40% of the cases to pulmonary lesions that can lead to a fatal outcome.
During the patient’s last admission, progression of his respiratory aspect was slow because initially he required the use of non-invasive mechanical ventilation. During the early course of his illness it was a good choice because the patient was able to tolerate it for 1 week, but subsequently deteriorated clinically with a state of shock that contraindicated the non-invasive mechanical ventilation. It was then decided to carry out orotracheal intubation. The patient developed acute respiratory distress syndrome. On the other hand, although the clinic and the PICU could provide guidance, ideally an echocardiogram should have been performed to demonstrate that the edema presented by the patient was not cardiogenic in nature. The patient probably developed pulmonary hypertension, given the hypoxemia and the need for high frequency oscillatory ventilation as well as the need for milrinone with its pulmonary vasodilating properties as part of the inotropic management.
Hemodynamically the patient presented septic shock, requiring high doses of vasopressors and inotropes, evolving to multiple organ failure with involvement of the circulatory system manifested by a state of shock, respiratory failure evidenced by marked respiratory acidosis and increased pulmonary shunts as well as acute renal failure characterized by anuria, creatinine of 1.3 mg/dl and glomerular filtration rate of 43, perhaps combined with renal hypoperfusion secondary to hypovolemia due to the massive pulmonary bleeding.
With respect to the infectious management, initial coverage was prescribed only based on the diagnosis of severe pneumonia and not with a septic patient in mind. The sulbactam amoxicillin coverage spectrum was very similar to cefotaxime and probably the reason why the change was unsuccessful. Guidelines suggest in these cases the use of a β-lactam as empiric therapy, usually cefotaxime or ceftriaxone, in addition to an aminoglycoside, a treatment scheme that should have been the initial treatment. Due to the suspicion of a fungal infection in a septic patient, coverage with azoles should be indicated: this was correctly done. Within the context of the patient in septic shock, it would be logical to think that pulmonary hemorrhage addresses a consumptive coagulopathy. However, platelets, coagulation times and fibrinogen were all reported to be within normal ranges. Therefore, the cause of the pulmonary hemorrhage might have been pulmonary manifestations of Kaposi sarcoma or a paraneoplastic pemphigus, although these theories could not be confirmed in the absence of a skin biopsy.
In the analysis, it is difficult to determine if the patient’s signs and symptoms were as a result of the aggressive CD presentation or of an undiagnosed immunodeficiency (or both) and, therefore, untreated. It cannot be stated that this patient could have been saved, but early initiation of treatment for multicentric CD and raising the index of suspicion of acquired immunodeficiencies in these patients can be suggested.
Final diagnoses:
1. Mixed type or plasma cell CD
2. Probable secondary immunodeficiency
3. Kaposi sarcoma
4. Paraneoplastic pemphigus
5. Severe community-acquired pneumonia
6. Acute respiratory distress syndrome
7. Sepsis
8. Acute renal failure
9. Septic shock
10. Multiple organ dysfunction
Immediate cause of death: pulmonary hemorrhage.
3.2. Pediatrics (Dra. Paloma Real Castrejón)
Lymph nodes >1 cm are identified as lymphadenopathy. It is necessary to determine their classification as localized and generalized. Localized nodes are those found in the same region, and generalized nodes are those found in two non-contiguous regions. It is important to rule out that the most common etiology of lymphadenopathies is infectious. There are also the neoplastic type, immunological type, manifestations of endocrine diseases, and others classified as miscellaneous. Within this approach, the clinical history and the preliminary case history are important: duration of lymphadenopathies, contact with sick individuals, whether there is a complete vaccination scheme, and if there were recurrent infections and constitutional symptoms. During physical examination, size of the lymphadenopathy should be determined along with the location, infiltration to deep planes and consistency. This information would have pointed to the diagnosis. Based on the history obtained during the interview, certain complementary studies can be requested such as blood count, COOMBS, coagulation times, chest xray, serological tests for cytomegalovirus, Epstein-Barr virus, toxoplasmosis, Brucella spp., rheumatoid factor, anti-DNA and anti-ANA. Occasionally, computerized tomography (CT) helps to outline the extent of the adenopathy as well as adenomegalies that cannot be palpated such as mediastinal, mesenteric and retroperitoneal.
A biopsy should be performed in those adenopathies >3 cm and in those in a patient with constitutional symptoms as well as those nodes with a hard consistency that infiltrate to deep tissues and are located in the supraclavicular region. It is important to remember that these studies are not requested at the same time and the clinical interview and physical examination are the procedures that will point out which studies should be initially requested.
4. Pathology (Dr. Mario Perezpeña-Diazconti)
The first contact we had with this patient was on November 30, 2011 when we received a lymph node that was increased in size measuring 4 cm at its greatest axis (Fig. 4A). Histologically there is expansion of the paracortical area with large lymphoid follicles of variable size, increased in number and with concentric arrangement of lymphocytes, which formed what is described as onion skin, with active germinal centers (Fig. 4B). The vessels show hyperplasia of the wall and concentric proliferation of fibroconnective tissue (Fig. 4C). Immunohistochemical studies were done (CD20) in which the population of B cells in the germinal center (as it is normal in the lymph nodes) and of T-lymphocyte around these germinal centers were highlighted. No data of malignant neoplasm were observed. The patient was diagnosed with hyalinovascular CD.
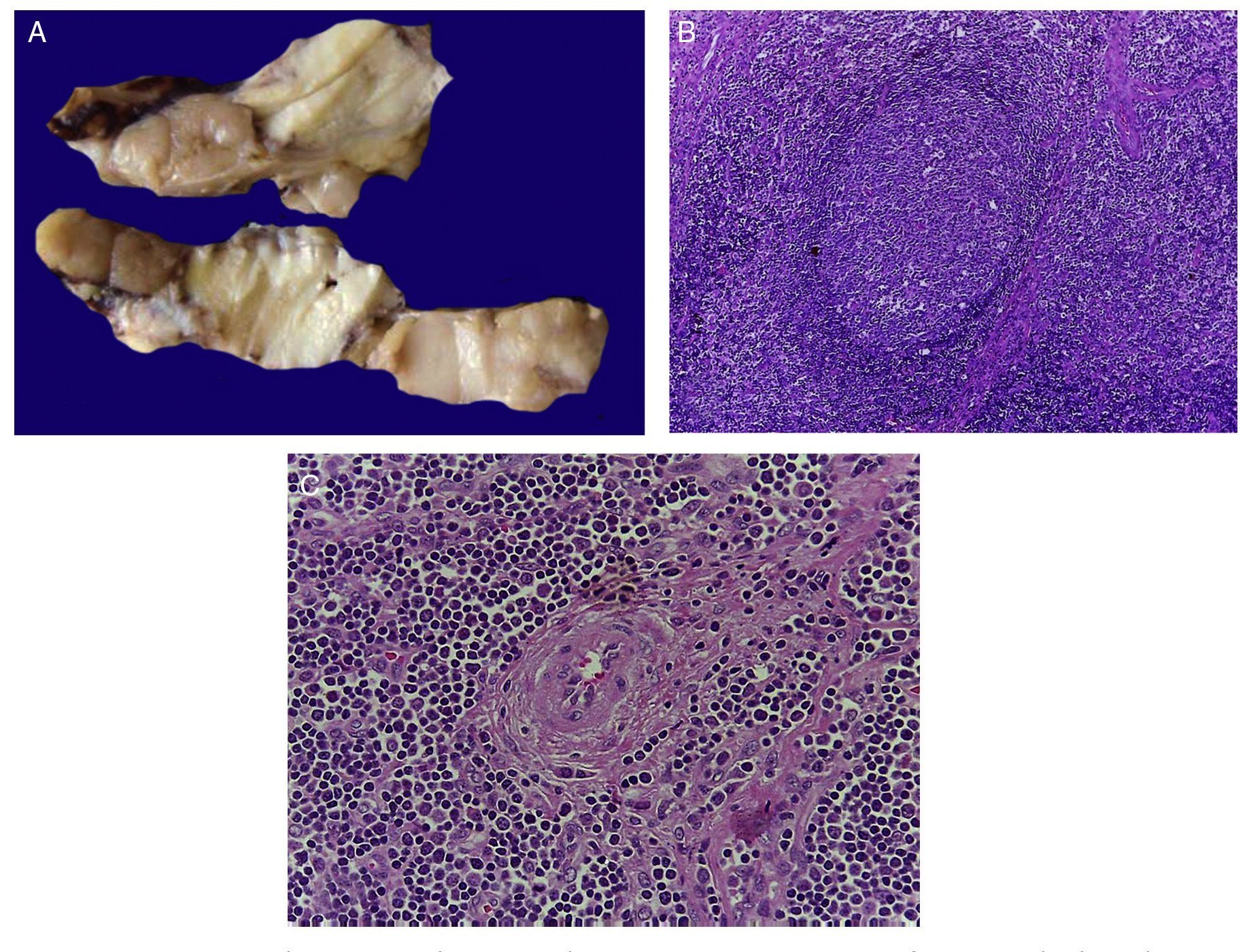
Figure 4 Castleman disease or hyperplasia of giant lymph nodes. Macroscopically, significant growth of lymph node cluster is shown. During autopsy, these measured 3-4 cm of the major axis of each (A). Follicular hyperplasia with concentric arrangement of peripheral lymphocytes (B). Vessels show hyperplasia of the wall (C).
Immunohistochemistry results were as follows: Cervical lymph node
• CD10: positive in germinal centers
• CD20: positive in the cortex
• CD3: positive in the paracortex
• LMP1: negative
• CD30: negative
• BCL-6: positive in the germinal centers
Subsequently, on December 28, 2011 another lymph node was received. The histological appearance was similar to the one previously received (Fig. 4). Confirmatory diagnosis was hyalinovascular CD.
February 23, 2012. Cytology from oral cavity vesicles was received. It was reported that there were inflammatory cells with poor preservation, changes due to drying and without viral inclusions. Once again, on March 09, 2012, another lymph node was received in which, as opposed to previous lymph nodes, there was loss of morphology as it was not possible to determine the follicular or the paracortex zones. A starry sky pattern was observed with macrophages with stained bodies. Immunohistochemical reactions were done with positive CD20 in the germinal center. Areas of T-lymphocytes were positive for CD3 and, in this case, CD138 was also done to show the presence of plasma cells, present in the CD variety, which were very scarce. Diagnosis of diffuse paracortical hyperplasia was made, negative for neoplastic cells. A bronchial aspirate was then received with scarce inflammatory cells. No respiratory epithelium cells were seen or Aspergillus sp., pneumocystis or bacteria.
June 08, 2012. An esophageal biopsy was received with ulcerated areas of the mucosa with inflammatory infiltrate, fibrin and cellular debris. No microorganisms were observed. Diagnosis was acute ulcerated esophagus.
4.1. Autopsy
The patient presented malnutrition, ulcerated lesions and blisters on the lips as well as in the oral cavity (Fig. 5). No blistering lesions were seen on the body. He had poorly defined “café au lait” spots of variable size and which practically occupied the anterior side of the chest and abdomen. Upon opening the cavities, serosanguineous fluid was seen in the pleural cavities, 50 ml from each side. In the abdominal cavity 40 mL of serosanguinous fluid was found. Lymph node chains were very increased in size. Each node measured from 2-4 cm and involved all the lymph node chains, some with extensive necrosis. Histologically, there was complete necrosis of the lymph node or loss of the morphology without necrosis. Staining for histochemistry was carried out on the nodes to check for yeast or bacteria, with negative results.
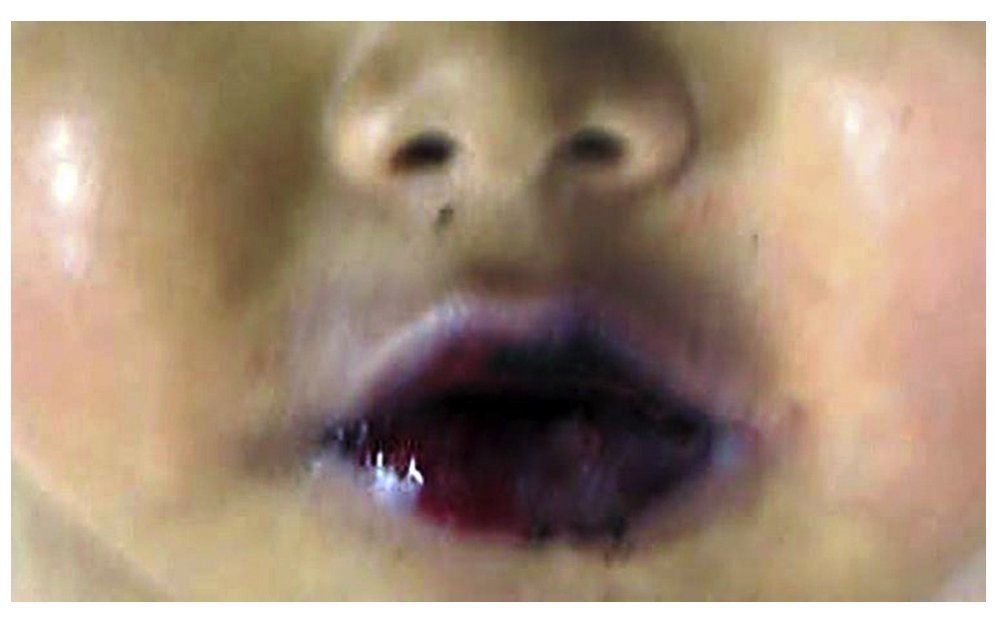
Figure 5 Lesions in the lips and oral mucosa showed blisters and ulceration of the mucosa.
In the trachea a large amount of cellular debris and a blood clot completely obstructing the lumen were found (Fig. 6A). The lungs weighed 250 g, with increase in weight and size. Histologically, the mucosa of the main bronchus showed ulcerated squamous epithelium with a large quantity of inflammatory infiltrate, cell debris and necrosis (Fig. 6B). In the pulmonary parenchyma there was bleeding and formation of microabscesses, formation of hyaline membranes due to multifactorial damage and focally multinucleated giant cells seen, suggestive of a viral infection (Fig. 6C). The spleen was increased in size and weight (Fig. 7A) with a nodular appearance and extensive areas of ischemic infarcts related to the shock or sepsis (Fig. 7B). The liver was increased in size and weight and congested histologically without other changes. The pancreas was increased in size and weight (Fig. 8A). Histologically, we found necrotic areas of the pancreas acinar (Fig. 8B). The esophageal and stomach mucosa showed ulcers of variable sizes. The largest affect the mid-third of the epithelium and lower third of the esophagus including the junction with the stomach (Fig. 9A). Histologically, the mucosa disappeared and was replaced by fibrin, cell debris and polymorphonuclear leukocytes and inflammatory infiltrate below the mucosa along with the presence of some multinucleated cells, suggesting the presence of some type of viral infection. This damaged extended up to the gastroesophageal junction and gastric mucosa. In the larynx, the mucosa was ulcerated with abundant inflammatory infiltrate and cells suggestive of a viral process. Also found was the presence of small spherical structures corresponding to the yeast Candida sp. (Fig. 9B). According to the clinical history there was decrease in the platelet count. In the bone marrow there was an increase in the number of megakaryocytes; however, they did not demonstrate morphological changes and no myelofibrosis was seen (Fig. 10). The principal diagnosis was vascular hyaline CD treated with steroids. The necrosis that was seen in the lymph nodes during the autopsy was due to the treatment administered.
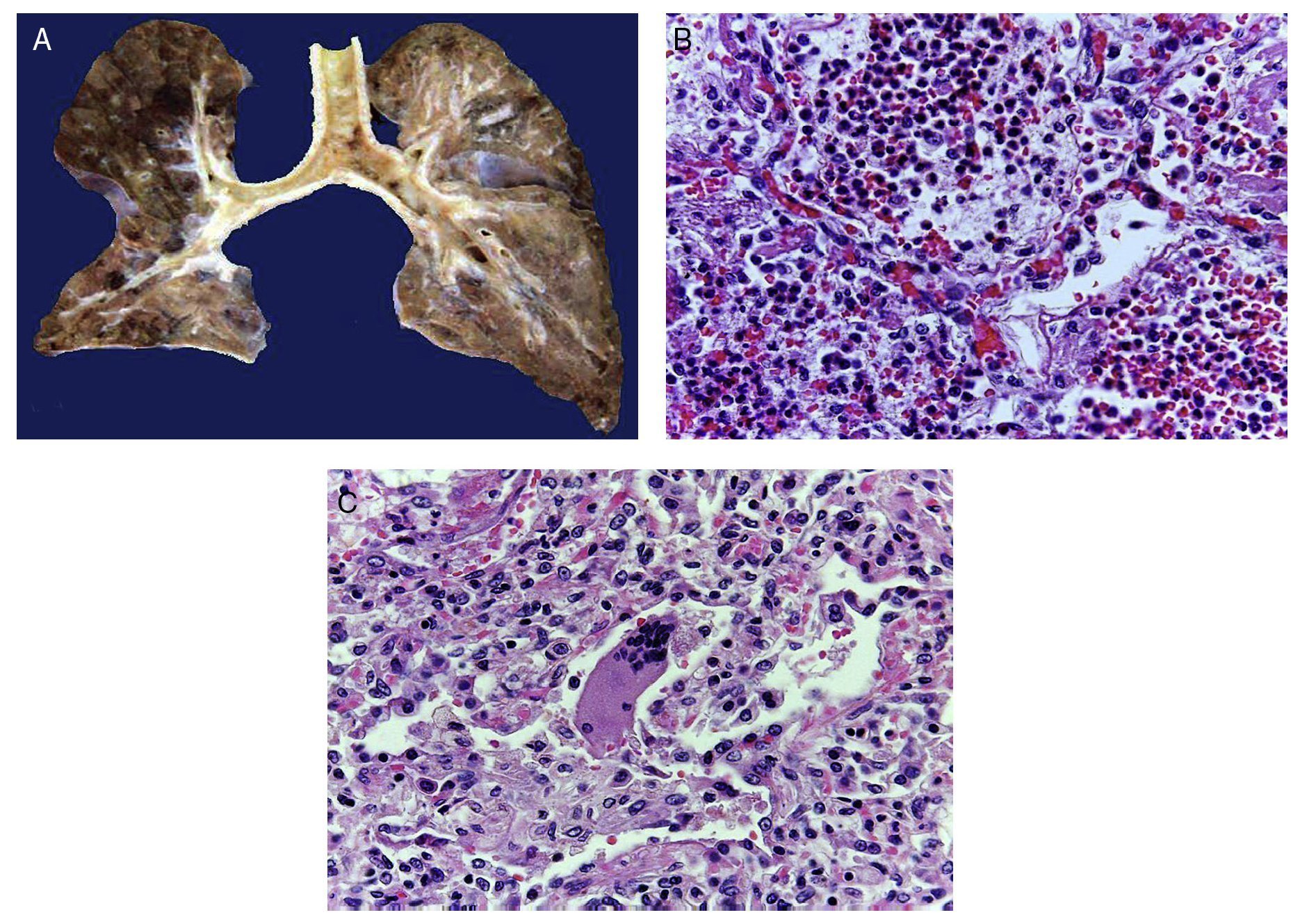
Figure 6 The tracheal lumen is occupied with fibrin, cell detritus and bloody material. When cut, the main bronchi show mucosal ulceration and the parenchyma is reddish brown with a solid appearance in hemorrhagic areas (A). Histologically, the alveolar spaces are occupied by polymorphonuclear leukocytes including abundant eosinophils and macrophages (B). In some areas there are giant multinucleated cells suggestive of viral infection (C).
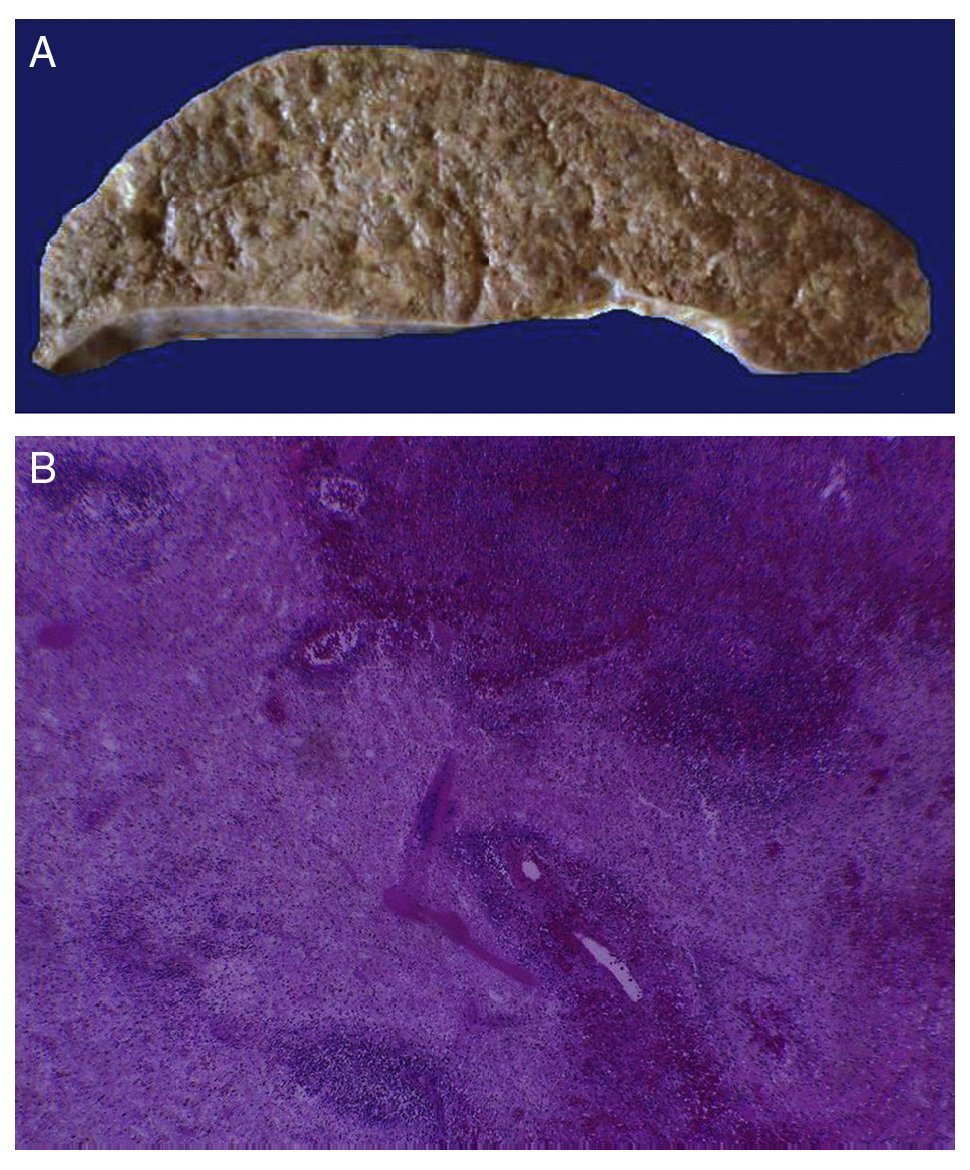
Figure 7 The spleen is increased in size and weight (A). Histologically, extensive zones of necrosis are observed associated with shock (B).
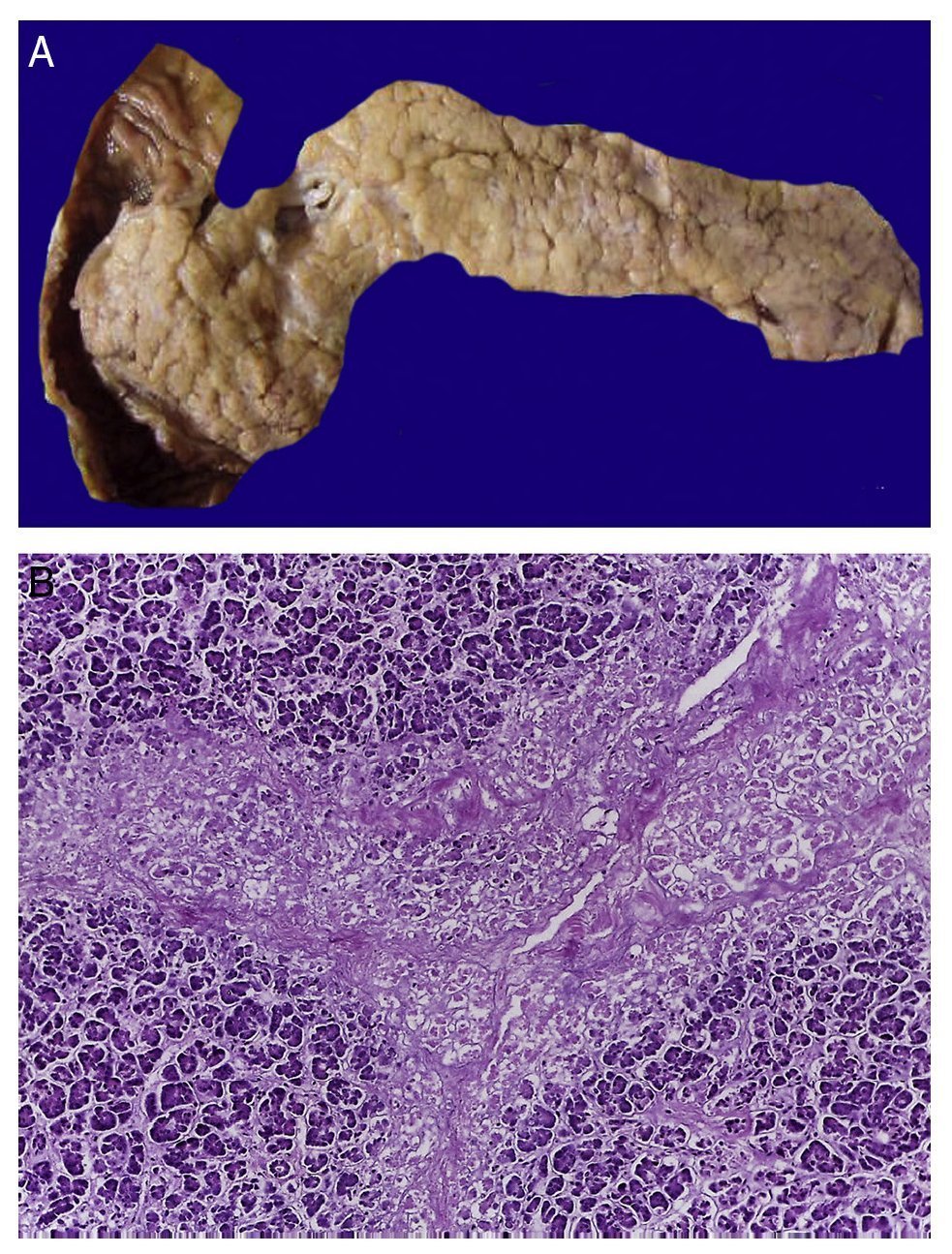
Figure 8 In the pancreas there is extensive necrosis of peripancreatic adipose tissue (A) and with an acinar component (B).
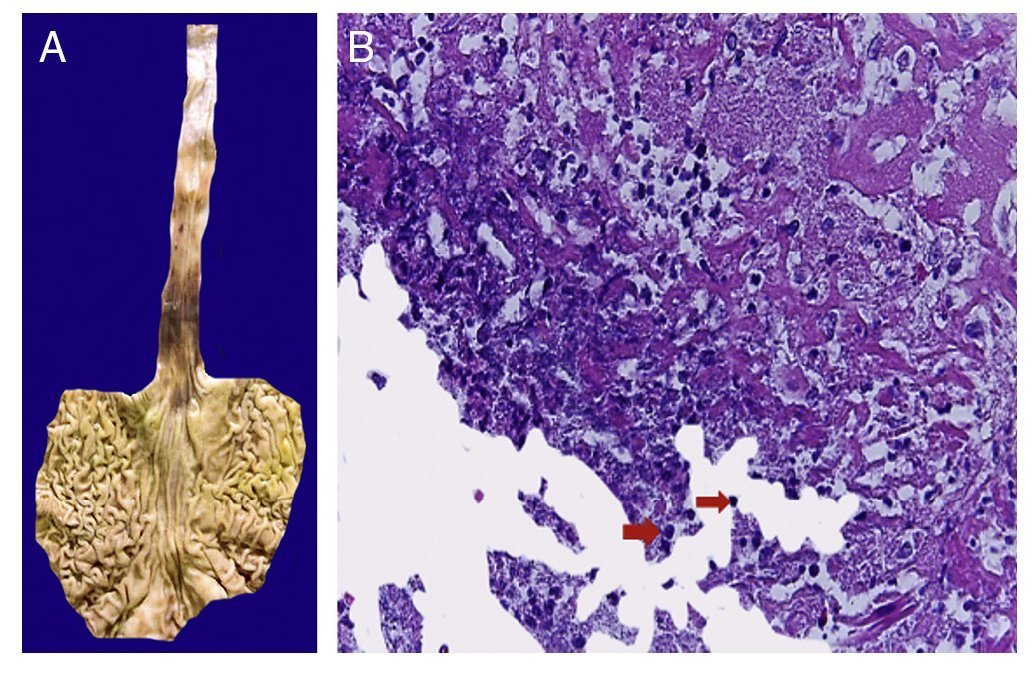
Figure 9 Stomach and esophagus. The mucosa of both show extensive ulceration affecting the mid-third and lower esophagus and gastroesophageal junction (A). Histologically, the ulcer is covered with fibrin, cell detritus and spherical structures that correspond to yeast of Candida sp. (arrows), and giant multinucleated cells, as observed in the lung (B).
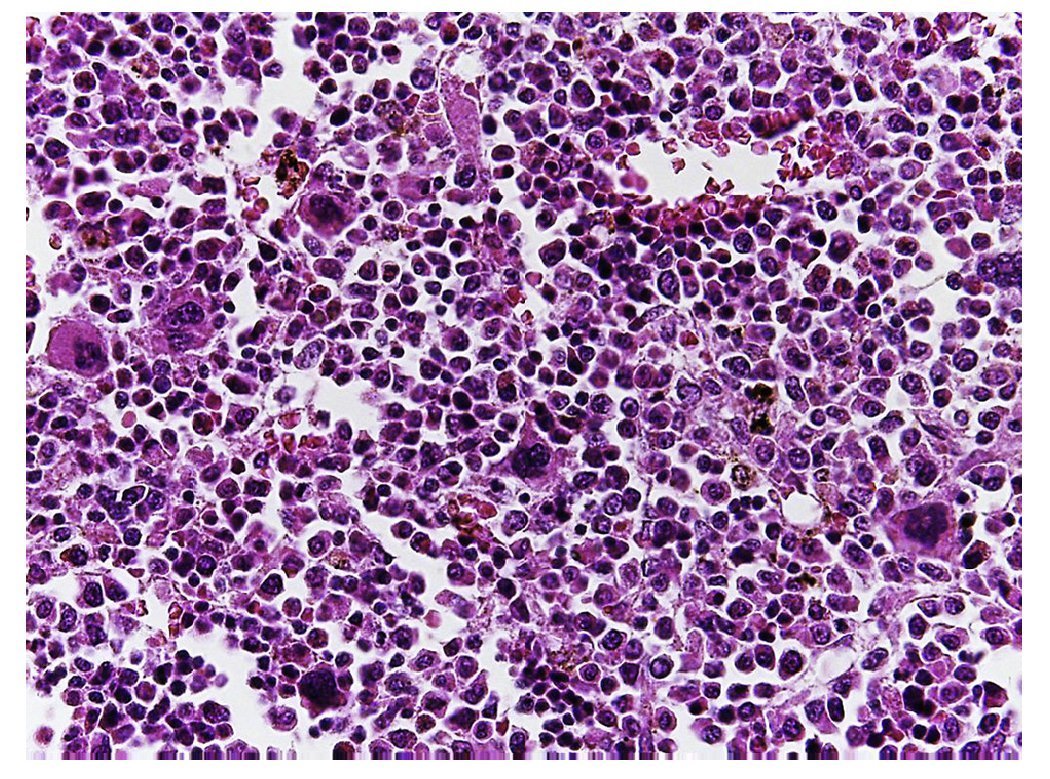
Figure 10 The bone marrow shows an increase in the number of megakaryocytes. However, their morphology is preserved and no myelofibrosis is observed.
4.2. Final anatomic diagnoses
Final anatomic diagnosies were vascular hyaline CD treated with steroids, with acute nonspecific necrotizing lymphadenitis CD involving the cervical, mediastinal, axillary, retroperitoneal and inguinal lymph nodes.6-10
4.3. Concomitant disorders
• Mixed acute bronchopneumonia with cytologic changes suggestive of a viral or bacterial infection
• Diffuse multifactorial alveolar damage due to sepsis and state of shock
• Extensive pulmonary hemorrhage
• Bilateral pneumothorax (50 mL right and left)
• Mucositis with glossitis and acute ulcerated laryngitis with cytological changes suggestive of a viral infection
• Acute ulcerated esophagitis affecting the mid-third and distal portion with cytological changes suggestive of a viral infection
• Congestive hepatomegaly (PO 1100 g/PE 588 g Kayser)
• Splenomegaly (PO 120 g/PE 58 g Kayser)
• Enlarged kidney (PO 180 g/PE 110 g Kayser)
• Lungs (PO 500 g/PE 275 g Kayser)
• Adrenal glands (PO 10 g/PE 7.5 g Kayser)
• Fluid in the peritoneal cavity (40 mL serosanguineous)
• Recent splenic infarcts
• Acute pancreatitis
4.4. Autopsy cultures
• Hemoccult, left and right culture of the lung, liver and intestine without bacterial growth
• CSF: scant growth of E. faecium
• Spleen: scant growth of E. faecium and Enterococcus sp.
• Colon: scant growth of E. faecium, Klebsiella pneumoniae and morphological types of E. coli CD presents two histological varieties: the hyaline vascular and plasma cells (multicentric). Similarly, it may have different clinical presentations: 1) single (can be a single node affected, and upon removal the patient is practically cured); (2) multicentric (severe form that almost always leads to death, which is what this patient presented).
Multicentric CD or idiopathic plasma lymphadenopathy is divided into two major groups with different clinical characteristics. As in this case, patients have pleural effusion, ascitis, systemic lymphadenopathy, thrombocytopenia and autoimmune diseases when they have polyclonal hyperimmunoglobulinemia. Patients without polyclonal hyperimmunoglobulinemia present with thrombocytopenia, anasarca/ fever, reticular fibrosis of the bone narrow, organomegaly, EC-HV and thrombocytopenia <100,000 platelets. According to what has already been stated, the patient presented was a carrier of a multifocal CD of the polyclonal hyperimmunoglobulinemia type.
5. Final comments
5.1. Coordinator (Dra. Martha Avilés Robles)
When CD is localized, surgical treatment resolves the disease. However, if the disease is multicentric, the response is generally inadequate and different maneuvers or treatment strategies are required.
5.2. Oncology (Dra. Elisa Dorantes Acosta)
Diagnostic suspicion is essential. One must think of very rare lymphoproliferative disease; in fact, its incidence is not exactly known in the literature. It is known that it is more common in adulthood, but the incidence in the pediatric age has not been adequately described.
Returning to the previous comments, if there is a lymph node of increased dimensions, if standard therapy with nonsteroidal antiinflammatory drugs (NSAIDs) was already administered without resolution, or if the node is in an area considered as pathological, a biopsy should be done. Biopsy should be excisional, especially in pediatric patients. This is very important so that pathology can observe the entire architecture of the lymph node. In reports of cases with CD there is the problem that when fine needle aspiration biopsy is done, usually a second excisional biopsy is needed. In fact, if the disease had been localized with only surgical excision of the lymph node, the child would have been cured. However, in this case in which the disease was multi-centric or systemic, several considerations should have been made:
1. Diagnosis of CD is made by pathology. When there is suspicion of an abnormal lymph node, the pathologist can provide the diagnosis of CD because differentiation will be made between a malignant process and this type of process which, although it was not cancer, was associated with several characteristics. This is not an isolated disease; therefore, immunodeficiency should be considered in association with some type of virus, etc. Viral serologies should be done when there is a report of CD in order to be able to make a precise diagnosis.
2. If the disease is systemic, first it must be determined whether any of the lymph nodes are causing problems that place the patient at risk. There are descriptions of patients with extensive mediastinal masses that cause respiratory or circulatory problems in whom urgent steroid therapy should be started.8 In other reports it is even considered to give localized radiotherapy to those lymph nodes that may place the patient’s life at risk, even if it is a systemic disease.
Once an evaluation has been done to see if there is risk of any lymph node that is compromising the life of the child independent of CD, consideration should then be given to what is going to be tried as a first approach to treatment. There are no guidelines available for management of this disease. The experience reported is of two isolated cases: use of prednisone has been tested for 2 months and later the reduction with monoclonal antibodies with fair results. On the other hand, therapy with vinca alkaloids has been tried. Likewise, it is recommended to surgically remove the lymph node causing the most effect and to then radiate the area. Therapy is diverse as there is no international agreement. However, it could be resolved according to a pediatrician using the approach of a diseased lymph node.
It was taken into consideration that this patient had a systemic disease of the hyalinovascular type, and other factors that could be associated were ruled out. However, the syndromatic POEMS group was not ruled out: Polyradicular neuropathy, which was not included in the clinical summary but could have been directly asked; Organomegaly, which the patient did not have, Endocrine disease, which was not asked; Monoclonality, which the patient did not have for at least one lymphoma; and Skin disorders, which he did have. The patient had some data probably related to this syndromatic group of multiple conditions, which were not ruled out. Due to the rarity of the diagnosis, it should have been considered as a differential diagnosis and these other disorders that are not mentioned in the clinical summary ruled out, which could have been corrected. The possibility of the child having a primary immunodeficiency was not examined in depth, although he did not have a secondary one (HIV). However, this approach was necessary to carry out the diagnostic group and probably offer some other type of treatment.
5.3. Coordinator (Dra. Martha Avilés-Robles)
During the course of his illness this patient developed a paraneoplastic pemphigus, which was not suspected until the disease was at a late stage. It is important to remember its form of presentation in order to have a timely diagnostic suspicion.
5.4. Dermatology (Dr. Carlos Mena-Cedillos)
Paraneoplastic pemphigus is a rare disease of the lymph nodes and related tissues. Although it is not cancer, it is considered to be a lymphoproliferative disorder. Some multicentric forms can behave aggressively and require treatment with chemotherapy and/or radiotherapy.11,12
Dermatology participation is not actually so direct in terms of general symptoms. It was noted at the time of the admission that the child had very nonspecific manifestations. In fact, it was a papular scaly erythematous dermatosis, which in reality is not a diagnostic description per se. It was approached as a reactive dermatosis with various possible lesions as reported in the literature: flaccid or tense blisters, with or without erosion (on the patient during his second admission these were noted to be necrotic), with erythema multiforme and lichen planus-type lesions. This polymorphism may be present in the same patient at the same time (such as in this case), as well as occurring throughout his evolution. During the first admission the patient presented with scaly erythematous lesions and on the second admission the blister lesions were evident, with the addition of stomatitis.
In children, it is more common to find lichenoid dermatitis on the trunk and limbs, unlike what is mentioned in the adult, which was what was observed in the child’s first admission. It is even common to find palmoplantar compromise and nail involvement.
In terms of the paraneoplastic pemphigus, it is perhaps the most relevant part of the dermatological expression. Unfortunately, in this case it was not able to be documented. Oral mucosa compromise is a consistent finding and is, in fact, the first sign of the disease. The erosions and painful ulcerations that affect the mucosa and extend to the vermilion are characteristic, difficult to manage and refractory to treatment. There is also a compromise of the conjunctivae, esophageal and anogenital mucosa.
The following are the Anhalt criteria of paraneoplastic pemphigus12:
• Clinical: painful mucosal erosions with polymorphous skin lesions associated with a neoplasm
• Histological findings: intraepidermal acantholysis, necrotic keratinocytes, vacuolar interface dermatitis
• Direct immunofluorescence: deposits of complements and IgG in the intercellular spaces of the epithelium and a variable manner throughout the basal membrane
• Indirect immunofluoresence: presence of circulating antibodies that recognize the intercellular spaces of the transitional and stratified epithelium
• Immunoprecipitation of serum antibodies against high molecular weight antigens: desmoplakin 1 (250 kD), bullous pemphigoid antigen (230 kD), desmoplakin 2 and envoplakin (210 kD), periplakin (190 kD) and an unidentified antigen of 170 kD
The Camisa and Helm diagnostic criteria include three major criteria: polymorphous mucocutaneous rash, associated internal neoplasm, characteristic immunoprecipitation test. Three lesser criteria are as follows: acantholysis in histopathology, direct perilesional immunofluorescence with intracellular pattern and in the area of the basal membrane, positive indirect immunofluorescence on rat bladder. The three major criteria or two major and two lesser criteria must be met.13
In the publication by Cervini et al. there is reference made to a 10-year old female with CD and paraneoplastic pemphigus and another reference to a 12-year-old male with paraneoplastic pemphigus associated with Hodgkin’s lymphoma.13 It is very important that when the physician is faced with a patient with CD with paraneoplastic pemphigus that the differential diagnoses be carried out, especially with diseases that have refractory stomatitis, in this case due to confirmation of the presence of Candida in the mucosa. This may have delayed the diagnosis of paraneoplastic pemphigus due to its expression at the level of the mucous membranes, but it must also be distinguished from erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, pemphigus vulgaris not associated with neoplasms, bullous pemphigoid, lichen planus, herpes infections, among others.
This case could have been demonstrated through immunological tests.
Ethical responsibilities
Protection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of Data. The authors declare that no patient data appear in this article.
Right to privacy and informed consent. The authors declare that no patient data appear in this article.
Conflict of interest
The authors declare no conflict of interest of any nature.
Received 4 May 2015;
accepted 6 May 2015
http://dx.doi.org/10.1016/j.bmhimx.2015.05.005
* Corresponding author.
E-mail:martha.aviles15@gmail.com (M. Avilés-Robles).