










Acute lymphoblastic leukemia (ALL) is the most common childhood cancer. It is well-known that genetic alterations constitute the basis for the etiology of ALL. However, genetic abnormalities are not enough for the complete development of the disease, and additional alterations such as epigenetic modifications are required. Such alterations, like DNA methylation, histone modifications, and noncoding RNA regulation have been identified in ALL. DNA hypermethylation in promoter regions is one of the most frequent epigenetic modifications observed in ALL. This modification frequently leads to gene silencing in tumor suppressor genes, and in consequence, contributes to leukemogenesis. Alterations in histone remodeling proteins have also been detected in ALL, such as the overexpression of histone deacetylases enzymes, and alteration of acetyltransferases and methyltransferases. ALL also shows alteration in the expression of miRNAs, and in consequence, the modification in the expression of their target genes. All of these epigenetic modifications are key events in the malignant transformation since they lead to the deregulation of oncogenes as BLK, WNT5B and WISP1, and tumor suppressors such as FHIT, CDKN2A, CDKN2B, and TP53, which alter fundamental cellular processes and potentially lead to the development of ALL. Both genetic and epigenetic alterations contribute to the development and evolution of ALL.
La leucemia linfoblástica aguda (LLA) es el tipo de cáncer más frecuente en niños. Aunque se sabe que las alteraciones genéticas constituyen la base de la etiología de la LLA, se ha demostrado que no son suficientes para el desarrollo leucémico; son necesarias alteraciones adicionales, como las modificaciones epigenéticas. En la LLA se han identificado alteraciones de este tipo, como la metilación del DNA, la modificación de histonas y la regulación por RNAs no codificantes. La hipermetilación del DNA en regiones promotoras es una de las alteraciones epigenéticas más frecuentes en LLA: conlleva al silenciamiento de genes que generalmente son supresores de tumor y, en consecuencia, contribuye a la leucemogénesis. También se han detectado alteraciones en proteínas remodeladoras de histonas, como la sobreexpresión de enzimas desacetilasas de histonas, así como alteraciones en enzimas acetil transferasas y metil transferasas. En la LLA también se altera la expresión de miRNAs, lo cual produce desregulación en la expresión de sus genes blanco. Estas modificaciones epigenéticas son eventos clave en la transformación maligna, e involucran la desregulación de oncogenes como BLK, WNT5B y WISP1 y de supresores de tumor como FHIT, CDKN2A, CDKN2B y TP73, lo que afecta diversos procesos celulares fundamentales que conllevan al desarrollo de LLA. Las alteraciones epigenéticas y genéticas contribuyen en conjunto al desarrollo y evolución de la LLA.
Leukemias are the most common pediatric neoplasms; acute lymphoblastic leukemia (ALL) is the most frequent type.1 Diverse specific genetic alterations have been identified in ALL that lead to the development of this disease.1,2 However, it is well recognized that these are not sufficient for leukemic transformation. Therefore, additional events are required.3 Currently, it is known that—in addition to genetic alterations—epigenetic changes are also fundamental in the leukemogenic process.4–6
Epigenetics studies heritable changes that lead to modifications in gene expression, which not imply alterations to the DNA sequence.7,8 DNA methylation, histone modification, nucleosome remodeling and regulation of noncoding RNA are among the most studied epigenetic mechanisms (Figure 1).9,10 These epigenetic alterations constitute fundamental events in the development of ALL. In this review, the main epigenetic alterations found in ALL are presented and defined. DNA methylation, histone modification like acetylation, microRNA regulation, and genes modified by these mechanisms are included. Although epigenetic alterations are fundamental in ALL of B (ALL-B) and T (ALL-T) lineage in both adults and children, this review is focused mainly on pediatric ALL-B due to the high incidence of this disease in this age group. The study of epigenetic modifications in ALL is of vital importance for knowledge concerning the etiology of the disease, as well as for the search of potential therapeutic targets.4,9
2Acute lymphoblastic leukemia, histone modification (b), and regulation by non-coding RNAs (c). In each case, the conditions under gene expression is induced (green) or silenced (red) are indicated.")
ALL is characterized by the uncontrolled proliferation of immature lymphoid cells called lymphoblasts, which predominate in the bone marrow and alter normal hematopoiesis.2,11 ALL affects T or B lineage lymphocytes and occurs in adults and children.11 It is of special relevance in the pediatric stage since it is the most common cancer, representing 25% of the cases. Eighty-five percent of ALL cases in children are of the B precursor type (ALL-B). Pediatric ALL cases are phenotypically and genetically heterogeneous and the etiology has not been completely determined.12
ALL has been classified in subgroups according to the presence of genetic alterations, which have a strong influence on the patient's prognosis.13 Approximately 75% of pediatric ALL cases present some type of numerical or structural chromosomic alteration.14 The most common numeric alteration in pediatric ALL is hyperdiploidy15; translocations are the most frequent chromosomal structural alterations16. The t(12;21)(p13;q22) translocation generates the fusion of ETV6-RUNX1 genes and is present in 20-25% of the cases.15,16 The t(1;19)(q23;p13) translocation leads to the fusion of TCF3-PBX1 genes and is present in 5-6% of the cases of pediatric ALL.13 The t(9;22)(q34;q11) translocation generates the fusion of BCR-ABL1 genes and is present in 3% of the cases.11,16 Translocations involving the MLL gene (KMT2A) are detected in 3% of patients between 2-5 years of age and 80% of those under one year of age. Although these genetic alterations are capable of starting the process of leukemogenesis, they are generally not sufficient; hence, additional genetic or epigenetic modifications are necessary for the development and evolution of ALL.16
3DNA methylationDNA methylation is the most studied epigenetic mechanism in ALL.4 This modification involves the covalent addition of methyl groups to the carbon 5 of the DNA's cytosine. In mammals, DNA methylation occurs primarily in the sequence 5′-CpG-3′, known as CpG island.17 CpG dinucleotides are found in low density throughout the genome; however, there is a high density of this sequence in the promoter regions.17 Approximately, 70% of the promoter regions in mammals present CpG islands.10 DNA methylation of the promoting regions generally correlates with gene expression silencing.17 On the contrary, CpG methylation along the body of the gene promotes elongation during transcription.8
The enzymes responsible of adding the methyl groups to the DNA are known as DNA methyltransferases (DNMTs), and at least three have been discovered in eukaryotes.18 The DNMT1 is in charge of maintaining DNA methylation during replication: it methylates newly synthesized CpG dinucleotides.10,18 The DNMT3a and DNMT3b establish, primarily, de novo methylation during embryogenesis8,10; additionally, they repair errors by the DNMT1 during DNA synthesis.8 In contrast, demethylation of the DNA involves the enzyme TET2 (ten-eleven-translocation 2), which catalyzes the conversion of 5-methylcytosine to 5-hidroxymethylcytosine.19
DNA methylation is one of the most studied epigenetic modifications in cancer. In some types of cancer, tumor cells show a global hypomethylation of the DNA, which is associated with chromosomal instability, transposon reactivation, and loss of the genomic imprint.4,8 In general, cancer cells present hypermethylation of the DNA in promoter regions. It has been reported that abnormal methylation in cancer is present in 5-10% of the normally demethylated promoters.10 This abnormal hypermethylation can lead to silencing of tumor suppressor genes, and consequently to a neoplastic process.18 Abnormal hypermethylation not only affects coding gene expression but also noncoding RNA expression, which can contribute to malignant transformation.10 Alteration of DNA methylation is a highly prevalent event in several types of cancer, including ALL (Figure 2).
3.1Alterations in DNA methylation in ALL3.1.1Directed studies
Numerous studies have established that disruption of DNA methylation has a fundamental role in the development of ALL in both B and T lineages in adults and children. The study of the alterations in gene methylation in ALL has been approached in two ways: through the directed study of specific genes preselected for their importance in ALL and through genome wide studies in which the analysis is not limited to certain genes.20 In most cases, the state of gene methylation has been tied with studies of gene expression. Altogether, these studies have revealed aberrant methylation and expression, mainly simultaneous hypermethylation in multiple genes, which has been defined as a “methylator phenotype”. This alteration leads to gene silencing and is a common characteristic of ALL and other types of leukemia in adults and children.4,20,21
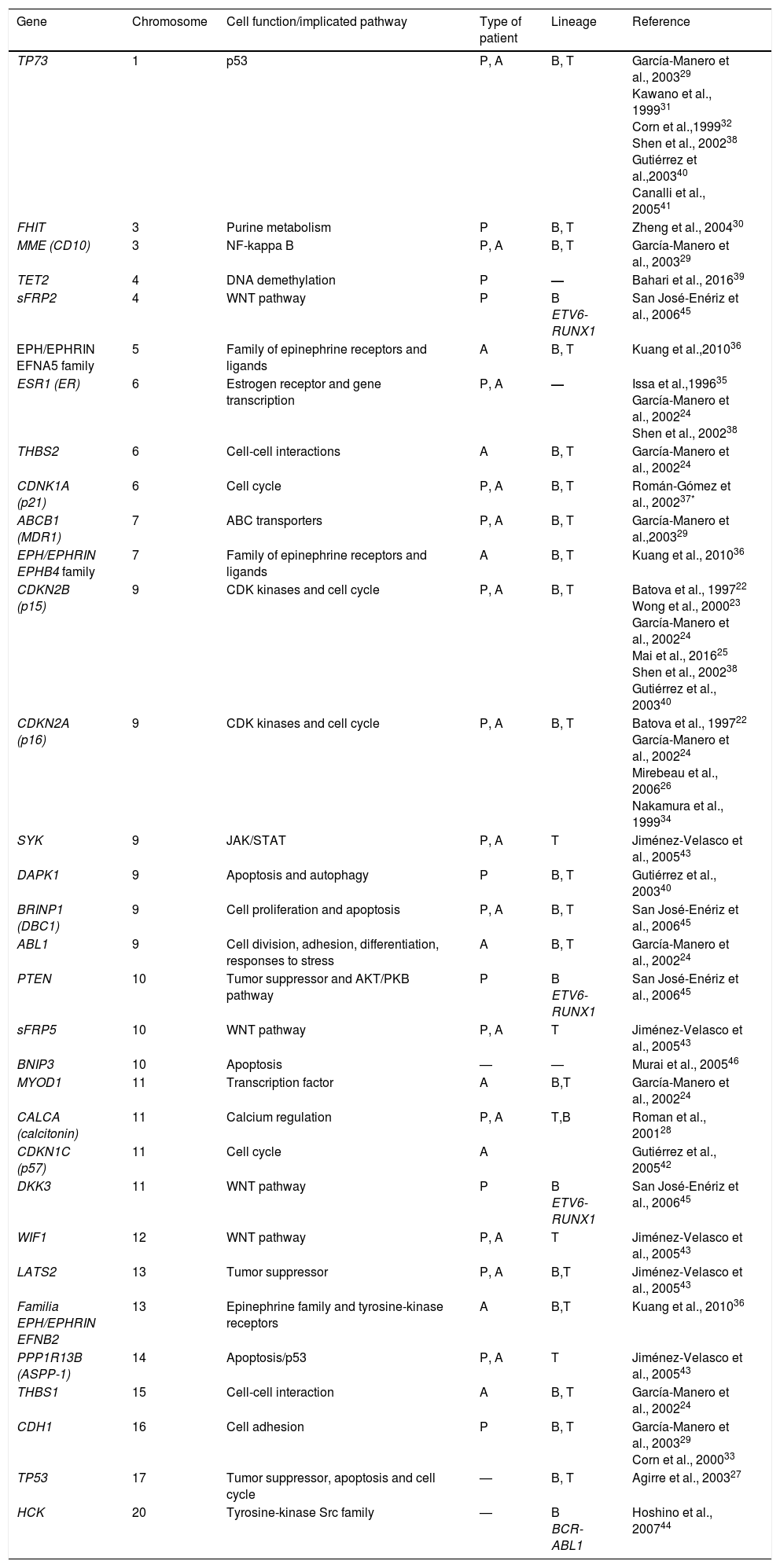
Through directed studies, several hypermethylated genes have been found in patients with ALL (Table 1). Some of these include genes such as TP73, FHIT, MME (CD10), TET2, sFRP2, EFNA5, ESR1, THBS2, CDKN1A (p21), ABCB1, EPHB4, CDKN2B (p15), CDKN2A (p16), SYK, DAPK1, BRINP1 (DBC1), ABL1, PTEN, sFRP5, BNIP3, MYOD1, CALCA (calcitonin), CDKN1C (p57), DKK3, WIF1, LATS2, EFNB2, PPP1R13B (ASPP1), THBS1, CDH1, TP53 and HCK.22–46 Most of these tumor suppressor genes are involved in cellular functions and fundamental signaling pathways, such as those regulating the cell cycle, apoptosis, DNA-damage response, and gene expression (Table 1). Based on the results of these directed studies, several correlations have been observed between the patterns of methylation and the subtypes of ALL have been obtained, as well as the prognosis of the disease.
Hypermethylated genes in acute lymphoblastic leukemia, identified through directed methylation studies.
| Gene | Chromosome | Cell function/implicated pathway | Type of patient | Lineage | Reference |
|---|---|---|---|---|---|
| TP73 | 1 | p53 | P, A | B, T | García-Manero et al., 200329 Kawano et al., 199931 Corn et al.,199932 Shen et al., 200238 Gutiérrez et al.,200340 Canalli et al., 200541 |
| FHIT | 3 | Purine metabolism | P | B, T | Zheng et al., 200430 |
| MME (CD10) | 3 | NF-kappa B | P, A | B, T | García-Manero et al., 200329 |
| TET2 | 4 | DNA demethylation | P | — | Bahari et al., 201639 |
| sFRP2 | 4 | WNT pathway | P | B ETV6-RUNX1 | San José-Enériz et al., 200645 |
| EPH/EPHRIN EFNA5 family | 5 | Family of epinephrine receptors and ligands | A | B, T | Kuang et al.,201036 |
| ESR1 (ER) | 6 | Estrogen receptor and gene transcription | P, A | — | Issa et al.,199635 García-Manero et al., 200224 Shen et al., 200238 |
| THBS2 | 6 | Cell-cell interactions | A | B, T | García-Manero et al., 200224 |
| CDNK1A (p21) | 6 | Cell cycle | P, A | B, T | Román-Gómez et al., 200237* |
| ABCB1 (MDR1) | 7 | ABC transporters | P, A | B, T | García-Manero et al.,200329 |
| EPH/EPHRIN EPHB4 family | 7 | Family of epinephrine receptors and ligands | A | B, T | Kuang et al., 201036 |
| CDKN2B (p15) | 9 | CDK kinases and cell cycle | P, A | B, T | Batova et al., 199722 Wong et al., 200023 García-Manero et al., 200224 Mai et al., 201625 Shen et al., 200238 Gutiérrez et al., 200340 |
| CDKN2A (p16) | 9 | CDK kinases and cell cycle | P, A | B, T | Batova et al., 199722 García-Manero et al., 200224 Mirebeau et al., 200626 Nakamura et al., 199934 |
| SYK | 9 | JAK/STAT | P, A | T | Jiménez-Velasco et al., 200543 |
| DAPK1 | 9 | Apoptosis and autophagy | P | B, T | Gutiérrez et al., 200340 |
| BRINP1 (DBC1) | 9 | Cell proliferation and apoptosis | P, A | B, T | San José-Enériz et al., 200645 |
| ABL1 | 9 | Cell division, adhesion, differentiation, responses to stress | A | B, T | García-Manero et al., 200224 |
| PTEN | 10 | Tumor suppressor and AKT/PKB pathway | P | B ETV6-RUNX1 | San José-Enériz et al., 200645 |
| sFRP5 | 10 | WNT pathway | P, A | T | Jiménez-Velasco et al., 200543 |
| BNIP3 | 10 | Apoptosis | — | — | Murai et al., 200546 |
| MYOD1 | 11 | Transcription factor | A | B,T | García-Manero et al., 200224 |
| CALCA (calcitonin) | 11 | Calcium regulation | P, A | T,B | Roman et al., 200128 |
| CDKN1C (p57) | 11 | Cell cycle | A | Gutiérrez et al., 200542 | |
| DKK3 | 11 | WNT pathway | P | B ETV6-RUNX1 | San José-Enériz et al., 200645 |
| WIF1 | 12 | WNT pathway | P, A | T | Jiménez-Velasco et al., 200543 |
| LATS2 | 13 | Tumor suppressor | P, A | B,T | Jiménez-Velasco et al., 200543 |
| Familia EPH/EPHRIN EFNB2 | 13 | Epinephrine family and tyrosine-kinase receptors | A | B,T | Kuang et al., 201036 |
| PPP1R13B (ASPP-1) | 14 | Apoptosis/p53 | P, A | T | Jiménez-Velasco et al., 200543 |
| THBS1 | 15 | Cell-cell interaction | A | B, T | García-Manero et al., 200224 |
| CDH1 | 16 | Cell adhesion | P | B, T | García-Manero et al., 200329 Corn et al., 200033 |
| TP53 | 17 | Tumor suppressor, apoptosis and cell cycle | — | B, T | Agirre et al., 200327 |
| HCK | 20 | Tyrosine-kinase Src family | — | B BCR-ABL1 | Hoshino et al., 200744 |
Genome wide association studies include an extensive analysis of the genome, as well as a greater number of patients.17,20 It is noteworthy that an association between directed studies and great scale analysis has not been found in all cases, possibly due to significant variations in the methodology and the size of the studied population.20
Genome wide association studies have compared samples of patients with ALL-B and leukemic cell lines with non-leukemic tissues and identified numerous changes in DNA methylation, mainly hypermethylation, in all subtypes of ALL-B.47,48 Some studies have focused in pediatric ALL or adult ALL and others have analyzed both groups. In most of these studies, patients with ALL-B and ALL-T have been included. Taylor et al. studied a limited number of patients, six children and four adults with ALL-B or T, and identified 11 genes (DCC, DLC1, DDX51, KCNK2, LRP1B, NKX6-1, NOPE, PCDHGA12, RPIB9, ABCB1, SLCA1) with aberrant methylation (Table 2) in comparison with normal cells of peripheral blood and bone marrow origin. In this study, no differences in methylation between ALL of B and T lineage were found, except in gene DDX51, which did not show methylation in ALL-T. Moreover, no differences between adults and children were found.49
Altered methylation/expression profiles in acute lymphoblastic leukemia identified through genome wide association studies.
| Study | Studied population | Lineage | Altered methylation genes |
|---|---|---|---|
| Taylor et al., 200749 | Adults: 4 Children: 6 | B and T | 11 hypermethylated genes: DCC, DLC1, DDX51, KCNK2, LRP1B, NKX6-1, NOPE, PCDHGA12, RPIB9, ABCB1, SLCA1 |
| Kuang et al., 200850 | Myeloid and lymphoid cell lines Adults: 61 | B and T | 15 hypermethylated genes: GIPC2, RSPO1, MAGI1, CAST1, ADCY5, HSPA4L, OCLN, EFNA5, MSX2, GFPT2, GNA14, SALL1, MYO5B, ZNF382, MN1 |
| Figueroa et al., 201352 | Children: 167 | B (137) and T (30) | 85 genes with alterations in methylation, common among all subtypes (73 hypermethylated, 12 hypomethylated*): TIE1, LPPR4, ANP32E, *DQX1, *AUP1, *HTRA2, *KCNJ3, GRM7, CEP97, DNAJC19, SENP2, DGKG, CETN3, CAMLG, PCDHA11, PCDHB17, PCDHB11, PCDHB11, PCDHGA5, PCDHGB3, *GNPDA1, PROP1, CMAH, *HIST1H2BJ, *HIST1H2AG, *HIST1H4I, SMAP1, FUT9, ATG5, SLC22A2, HOXA5, HOXA6, CHCHD2, BPGM, AGPAT6, MOS, SDR16C5, WDR67, WDYHV1, ELAVL2, C9orf25, DNAI1, MCART1, TAF3, KIAA1279, H2AFY2, NDUFB8, C10orf26, GPR26, CCDC34, *ELF5, LRRC4C, ZBTB16, SIAE, SPA17, TMTC1, C12orf44, SKA3, MRP63, DHRS12, OXA1L, PABPN1, SLC8A3, SCG5, TGFB1I1, CNOT1, SPNS2, ACACA, TADA2A, LSM12, G6PC3, *GPRC5C, OSBPL1A, INO80C, ZNF560, ZNF582, DDRGK1, ITPA, PTPRT, *OK/SW-cl.69, MAP7D2, GSPT2, PABPC5, MCTS1, MGC16121 |
| Chatterton et al., 201451 | Children: 69 | B | 370 genes with alteration in methylation (325 hypermethylated, 45 hypomethylated*) common among all subtypes: ZFP112, ZFP42, ZNF285, ZNF300, ZNF385D, ZNF677, ZFP37, ZNF560, CXCL1, CXCL3, CXCL5, CXCL6, ADRA1A, ADRA2C, AGTR1, CASR, FPR2, FZD9, GALR1, GPR126, GPR26, GPR37, GPR6, GPR77, GRM1, GRM7, HTR4, LHCGR, OPRM1, P2RY2, *PTAFR, PTGFR, QRFPR, SSTR1, GRIA2, GRIA4, GRIK1, GRIK2, GRIK3, KCNB1, KCNB2, KCNG1, KCNH7, KCNK1, KCNK5, KCNN2, *KCNQ4, TRPC1, TRPC4, TRPM3, SLC19A3, SLC1A1, SLC22A3, SLC24A3, SLC34A2, SLC44A4, SLC5A7, SLC6A1, SLC6A15, SLC6A2, PCDHB15, PCDHB2, PCDHB3, PCDH20, CDH6, CDH8, IL1RN, IL7, *LTA, ACADL, BHMT, CASD1, CYP24A1, CYP7B1, DHCR24, DPYS, DPYSL4, ELOVL4, EPHX2, GALNT11, GCNT2, GDA, LPL, LRAT, ME1, NMNAT3, *NOX3, PDE10A, PDE1C, PDE4B, PDE4C, PLOD2, PNPLA3, PRTFDC1, PTGS2, RGS7, SMPD3, ST6GAL2, UGGT2, CHI3L2, EFEMP1, KL, LYZ, PDZRN3, PLA2G7, POGLUT1, SMPDL3A, *DNTT, RFX6, ABO, ADCY2, *ENPP3, ENTPD3, GNA14, SH3GL2, MACROD2, HS6ST3, BMP7, FGF10, FGF12, GRP, KITLG, NELL1, PTN, NRG1, PEX5L, *VDAC3, CACNA1A, CACNA1D, CFTR, GABRA2, GLRB, GPM6A, HCN1, RYR2, NALCN, *BLK, *HSPB8, PRKD1, MYO3A, MAPK15, PAK7, *DDR1, EPHA5, ERBB4, FGFR2, KDR, NR2F2, PGR, THRB, *ARPP21, *EVL, *FGD2, *KISS1, *S100A2, *SORBS3, *SPRR4, *DEFB129, *MBP, *SFTPB, *WISP1, *WNT5B, *INTS1, *TCL1A, *CLDN15, *GP9, *LY9, *RPH3AL, *XIRP1, *ASB10, *FAM49A, *KLHDC7A, *SH2D4B, *SRBD1, ACTA1, DES, HSPA2, MT2A, NEFM, NEFH, SLPI, VSNL1, LIN7A, TOM1L1, CPLX2, RAB32, ERC2, PDLIM3, STXBP6, GULP1, PARVA, PARVG, NOD2, PREX2, CTTNBP2, RHPN2, PRKCDBP, SPATA18, SPTSSB, ADCYAP1, CBLN4, COL14A1, CYR61, FAM19A4, FAM20A, FSTL5, IGFBP3, LAMA1, LAMA2, MAMDC2, MATN2, NPTX2, RLN1, RLN2, SASH1, SCG3, SEMA3C, SERPINA1, SST, TAC1, TGFBI, TMEM45B, VSTM2A, ZPBP2, ACTL6B, CENPH, KIF6, MIPOL1, MLF1, NOL10, PRDM5, SNRPN, SYCP1, TDRD5, CADM1, CD8B, CDCP1, CLDN11, CNTNAP2, CTNND2, DSCAML1, EPB41L3, FLRT2, LSAMP, MARVELD3, NTM, PCDH8, PKP2, STEAP4, SYN2, THEM4, TPBG, ALS2CR11, C10orf107, C11orf70, C1orf115, C20orf197, C3orf14, C6orf118, C7orf63, CCDC37, CWH43, EFCAB1, FAM83A, FAM84A, FBXO39, FEZF2, FIBIN, LCA5, LPPR1, LRIG3, PHF21B, RBM47, SLC35F3, TMEM171, TMEM74, TSPYL5, TTC22, UNC80, WDR17, XKR6, CTSG, EPHX1, ADAM12, ADAMTS18, ADAMTS5, CPXM2, HP, PAPPA, PCSK1, PCSK2, PRSS12, TLL1, EYA4, PPP2R3A, CDC14B, PTPRO, PTPRZ1, LPPR4, ACTN2, BHLHE22, BNC1, DMRT1, DMRT2, DMRT3, EGR4, FOXA2, FOXG1, GATA4, GCM2, GSC2, HLF, HOXA2, HOXD1, HOXD11, ID4, IRF6, KLF1, *LIMD1, MSX2, MYF5, NACC2, NFE2, NFIB, NKX2-1, NPAS2, ONECUT2, OTX2, OVOL2, PAWR, *POU2AF1, POU4F3, PROX1, PRRX1, SCRT1, SIX1, SNCAIP, SOX1, SOX14, SOX17, SOX9, *TCF3, TLX3, TP73, WWC1, WWTR1, YAP1, ZIC1, EIF5A2, *ANXA9, CXADR, DCC, *ENG, FCGR2A, GPC6, *LRP8, *SCARF1, SFRP1, *HBZ, *SNX8, TF, UPF3A, SORCS3, ATP8A2, *LBP PDPN, SNAP25, SYT9, FAM164A, MOSC2 |
B, B cells; T, T cells.
Kuang et al. analyzed 23 cell lines of lymphoid and myeloid origin (MOLT4, Jurkat, Peer, T-ALL1, CEM, J-TAG, B-JAB, RS4, ALL1, Raji, REH, Ramos, K562, BV173, HL60, NB4, THP1, U937, ML1, OCI, HEL, MOLM13, KBM5R) and identified altered methylation in 65 genes. In 15 of these genes (GIPC2, RSPO1, MAGI1, CAST1, ADCY5, HSPA4L, OCLN, EFNA5, MSX2, GFPT2, GNA14, SALL1, MYO5B, ZNF382 and MN1) methylation was confirmed by analyzing 61 samples of adults with ALL-B and T (Table 2). The ontological analysis of these genes revealed enrichment of pathways involved in proliferation, cell growth and differentiation, apoptosis, gene expression regulation, DNA replication and repair, signal transduction, transport, and metabolism.50
In the study of Chatterton et al., in 2014, levels of expression and methylation in 69 children with ALL-B were associated. Hypermethylation and decreased gene expression in 325 genes was identified, as well as hypomethylation and increased gene expression in 45 genes. These results were compared with 42 non-leukemic bone marrow samples in which these modifications were not found (Table 2).51 Ontological analysis of these 370 genes showed greater representation of molecular pathways associated with hematologic neoplasms, such as cell to cell signaling and interaction, death and cell survival, as well as cell development. In particular, hypermethylation was found in genes codifying proteins involved in signaling of the glutamate receptor, G-protein signaling, and adenosine-monophosphate (cAMP) mediated signaling; suppression of several proteins involved in the cation transport was also found. Hypermethylation of these genes and its subsequent suppression are indicative of a reduction in the potential of cell-microenvironment interaction, as well as the apoptotic potential. In this study, alterations in methylation of genes associated with the leukemic process were also identified, such as chemokines CXCL1, CXCL3, CXCL5 and CXCL6 and cytokines LTA and IL-7, which genes showed hypo and hypermethylation, respectively. Additionally, 11 dysregulated kinase genes were identified: BLK, DDR1 and HSPB8, which showed hypomethylation and overexpression, and EPHA5, ERBB4, FGFRDZ, KDR, MAPKI5, MYO3A, PAK7 and PRKD1, which were found to be hypermethylated and, consequently, silenced. Alterations in factors of the WNT signaling pathway were also found: hypomethylation and overexpression of WNT5B and WISP1, as well as hypermethylation and low expression of SFRP1, PP2A, and SOX. The signature of methylation/expression was common in all ALL subtypes.51 This study also analyzed expression signatures dependent on the different subtypes of ALL and a comparison with non-leukemic samples was conducted. The study identified 55, 51, and 13 genes with epigenetic dysregulation associated with subtypes ETV6-RUNX1, hyperdiploidy and “other alterations” (this group included patients with fusion of TCF3-PBX1, BCR-ABL and MLL translocations), respectively (Tables 3–5).51
Methylation profile of the ETV6-RUNX1 subtype in pediatric ALL-B.
| Hypermethylated genes | Reference |
|---|---|
| NRG1, KCNC3, RAB37, COL7A1, VIPR2, NIPAL1, GREM1, TM6SF1, MTX2, DPP4, DOCK5, SYT17, WWTR1, ITGAX, TMEM144, ASNS, MYO3A, GUCY1B2, CHD5, ITGB5, PAX7, CD44, RAB6B, PI4K2A, ADRA1D, FAM123A, STBD1, OVOL2, RNF207, CRABP1, CDC42BPB, L3MBTL4, KIAA0020, MPZL3, PRICKLE1, ADPRH, PDCD1LG2, SLCO2A1, FXYD6, VAX2, GSX2, SYT12, TMEM67, SLAMF8, CTTN, LPXN | Chatterton et al., 201451 |
| MYO1E, STBD1, OPHN1, HOXD1, WWTR1, SOX3, SNAI3, ZNF462, C4orf31, LRRC55, ST6GALNAC4, C21orf15, ALX1, GUCY2D, FOXA2, ZNF462, SIGLEC9, MTMR11, RIN1, PDE3A, UCN2, KCNN2, NKX2-8, PDK4, NHS, HOXD1, TNFRSF9, NEFL, C20orf165, NEURL2, CTSA, BMP7, SLC35B1, ITIH4, MUSTN1, SLC2A5, MEFV, PLD4, CXCR3, DARC, C9orf79, STBD1, PDK4, UCN2, SRGN, DBX2, C10orf116, AGAP11, ZNF215, BGLAP, PCDHGB4, PCDHGA8, BMP7, PJA2, GPR50, ESX1, KRT80, ZNF804A, TFCP2L1, GNPNAT1, OGG1, ADCY3, CBLN4, CHAT, SLC18A3, PLEKHG4, WNT2, C20orf165, NEURL2, CTSA, GPR123, SYT15, FCN1, BDNF, FBXW5, C8G, C14orf49, EFEMP1, BBC3, LEPREL2, PCDHGB2, PCDHGA5, PCDHGB3, CRYBB3, PTMS, LAG3, FAM190A, EIF2C2, PCDHGA5, PCDHGB3, NEFM, CCBL1, LRRC8A, TUB, TJP1, LIMD1, PPARD, MGC16703, P2RX6, FBXW5, C8G, ZNF296, GEMIN7, GRIA3, NOS3, GABRE, RTDR1, RAB36, ZNF19, MSX2, TSEN2, PRDM11, EPHA5, GPR81, S100A9, EIF6, RPL9, LIAS, SCARF2, CUEDC1, DOK5, HECW1, GPR126, PRKAG3, MARCKSL1, HIST3H2A, HIST3H2BB, SIAE, SPA17, MAGEB6, C14orf49, DAAM2, EIF2C1, TSPAN15, MSRB3, ITGAE, HLF | Figueroa et al., 201352 |
| GRHL3, CTTN | Nordlund et al., 201553 |
| CLIC5, ACVR1C, IGF2BP1, DSC2, POLO | Lee et al., 201555 |
| Hypomethylated genes | |
| SLC35B3, EPOR, BEST3, APOBEC3A, MPP7, TMED6, TRIM69, ALPK1, MPP7, CRYBB3 | Chatterton et al., 201451 |
| MIB1, GFAP, TMED6, TPMT, KDM1B, GFAP, E2F6, DRD5, NCKAP1, CSTF3, HTR3C, HTR3C, FUCA1, DNAJC10, B4GALT3, BEST3, CALD1, SIDT1, LRRC4, GNB4, FCHSD2, ABHD3, RAD50, KCNK15, PIK3CG, LRRC4, C22orf24, YWHAH, RRAGA, NACA, VPS13C, SH2D3A, VAV1, C12orf49, RNFT2, ITSN2, TREML1, PRSS1, RAB3GAP1, MRPS15, ATF7, TNK1, ETV7, HLA-DQA1, ERLIN2, ST7L, CAPZA1, TUSC3, BAT2D1, LOC100127888, EID3, TSPYL1, DSE, ISG20, UFC1, ZNF143, SPEM1, C17orf74, C9orf40, MOBKL3, CAB39L, SETDB2, CSTF3, SLC35B3, ATP5J, GABPA, KCTD17, KLHDC5, TOR1AIP2, TOR1AIP1, CIITA, C12orf23, USP6NL, DHRS13, APOBEC3A, CCNI, TOPORS, EPM2AIP1, MLH1, FUT11, TIMM17B, PQBP1, SAA1, THOC7, ATXN7, SIRT2, ZCCHC3, PBXIP1, IRAK3, RAD51L3, FNDC8, TAP1, PSMB9, ISG20, ARHGAP24, PNRC1, DNAJC10, YARS, S100PBP, BTN3A2, RTP4, ELOVL5, DAXX, GAR1, LIMS2, GPR17, SERPINE1, YARS, S100PBP, ZNF644, POMP, RNF113A, NDUFA1, VWA5B2, CTCF, USHBP1, C19orf62, GAB1, C18orf56, TYMS, GABBR1, WDFY3, C4orf12, PRR5L, POLR3B, SPOP, VRK2, HABP4, NUP107, DLGAP2, ZNF566, C1orf59, TMEM127, CIAO1, RECQL4, LRRC14, HORMAD2, PIK3IP1, GLO1, CNR2, FAM71E1, C19orf63, TOR2A, BRWD1, SHE, TDRD10, CDC2L5, C21orf58, PCNT, MRE11A, ANKRD49, ERICH1, HLA-C, CHERP, TRIM9, CMTM2, CCDC62, HSPA8, SMC1B, RIBC2, PDE4DIP, ARHGAP25, USP49, GFI1, NOS1AP, UCP2, CLINT1, GGNBP2, RNF141, EXTL3, EXOSC9, SFT2D3, SEP15, HS2ST1, CACNA1I, ALG9, IKBIP, APAF1, SH2D4B, RAB33B, CLIP1, LOC344967, N4BP2, LOC100127888, LOC492303, ZNF326, FGFR4, HSPB11, LRRC42, HIST1H1C, ARF1, DHRS13, DSC3, SRGAP3, C6orf170, TAF6, CNPY4, MBLAC1, RAB37, CITED2, COG8, NIP7, NDUFA8, MORN5, SRP9, ZNF616, MTA3, FNBP4, GNGT2, ABI3, PELI3, ZNF423, CHDH, IL17RB, PPAT, PAICS, RASAL1, SRGAP3, CST8, ARF6, DIRAS2, LOC100128164, SEC62, MRPS36, FAIM, RAG2, C11orf74, USP50, GPR146, RAB3IL1, ARPP-21, HPS4, SRRD, UBE2J1, KRAS, C14orf142, UBR7, PTPRB, MAN1A2, VPS13B, MLL2, ZFP161, ZC3H7A, CD79B, EBF4, HNRNPK, RMI1, STRBP, OSGEP, APEX1, LOC100128164, SEC62, SMCHD1, SYNGR1, FLJ25006, LOC645851, SENP6, MAP1LC3A, C1orf156, C1orf112, CTHRC1, SCAP, C1QL4, ERCC6, ATPAF2, C17orf39, CALD1, PIK3C3, PAX6, WIPF1, COCH, KCNA3, MTMR10, RAB8B, CANT1, MANBA, POU5F1, RHBDL3, TIAL1, PIAS2, KPNA4 | Figueroa et al., 201352 |
| SESN2, E2F6, RP11-363D14.1, AC131097.3, XIRP1, ZDHHC3, RP11-803B1.2, SIDT1, RP11-71E19.2*, APBB2, KIF2A, DUSP1, RP11-274H24.1, TBC1D7, ZNF90P2, CUX1, ERICH1, ERICH1-AS1, ZNF704, FARP1, TMED6, CBFA2T3, IGF2BP1, AXIN2, MIB1, DSC3, EPOR, TCFL5 | Nordlund et al., 201553 |
| SOX11, SPSB1, BEST3, SIDT1, TCFL5, CHL1, FAM19A | Busche et al., 201359 |
Methylation profile of the hyperdiploid subtype in pediatric ALL-B.
| Hypermethylated | |
|---|---|
| MYO3A, WWTR1, VIPR2, PID1, FAM20A, SOX11, CRMP1, KL, WWC1, FGFR2, ZNF532, SLIT3, ENPP5, LOX, B3GALNT1, KIAA1456, ADAMTS1, AJAP1, DYNLRB2, AJAP1, MFSD7, DPY19L3, MAGI1, NRG1, PTPRG, FOXL1, FBN2, DSC3, FHIT, DSC3, FBN2, BMPR1B | Chatterton et al., 201451 |
| PPAT, PAICS, KCNF1, HORMAD2, FAIM, APPL1, DUSP15, TTLL9, LCK, CLSTN2, TXNDC11, CSDE1, ASB8, SETBP1, COL4A1, COL4A2, TEAD1, C1orf183, HIST1H1C, OIP5, NUSAP1, SCAP, VPS13B, ARHGAP25, SLC39A1, CREB3L4, RPS13, ABCB10, DSC2, POMP, SLC35B3, PDLIM3, WBP11, C12orf60, ACSM3, SEP15, HS2ST1, SCAP, NPR1, FAT1, INHBE, GLI1, ASAP2, BEST3, LOC145783, TCF12, TFAP2C, CNKSR2, PHLDB2, FBN2, EXTL3, HK2, CDC14B, C11orf30, RELN, TNIP1, HPS4, SRRD, USP11, GFI1, HNRNPK, RMI1, APOBEC3A, CST8, PBX3, ACACA, TADA2A, OSGEP, APEX1, C14orf142, UBR7, LOC100128164, SEC62, SRBD1, HLA-DQA1, PAX6, RAD50, ETV7, CHMP4C, MYOD1, CALD1, CSDA, CNTN2, KPNA4, GFAP | Figueroa et al., 201352 |
| Hypomethylated | |
| LOC284837, TNXB, C14orf49, LOC284837, NKG7, KRT36, ENTPD1, APOLD1, LSP1, DBNDD1, CLDN16, C14orf49, SEPT9, CHST15, BIN2, BCL2, BCL9L, CEBPG, VTCN1, CTNND1, KANK2, SLAMF1, LGALS3BP | Chatterton et al., 201451 |
| MX2, C14orf49, C14orf49, ACTR3C, LRRC61, SYNE1, FAM195A, WDR90, ARD1B, BCL3, SYNE1, SERPINA3, SERPINA4, CEP350, DEFA1, DEFA1B, DEFA3, NPBWR2, LRRTM4, ARID3A, C21orf15, ZNF366, MMP26, KRTAP12-1, SERPINA10, PARK2, PACRG, CLCA2, ARID3A, SERPINA4, FAM90A20, DCAF4L2, RAB24, PRELID1, SYN2, SH2D3C, PLEK, TRPT1, NUDT22, GEMIN4, ELP2P, SERPINA10, PNMA3, GPR56, XKR5, NFATC3, BGN, IL13RA1, LOX, TNXB, MX2, GP2, BGN, MAGEC2, AMT, NICN1, KL, KLF6, FAM92A3, ZDHHC21, VTCN1, NRBP2, ABCA12, GPR84, CRIP1, C9orf66, DOCK8, UBXN11, SH3D20, BTBD16, DARC, CYP2F1, ATP2B3, E2F2, B3GAT3, COASY, MLX, MAGEA12, CSAG1, CCT8L2, MMP12, NDUFS8, CLEC4D, C21orf84, KCP, EIF2C2, S100A13, S100A1, C1orf77, CLEC4D, FAM47A, FES, CALML4, COL1A1, PLEKHJ1, NAPRT1, MAGIX, FGD5, C7orf26, IFFO1, NCRNA00095, FBXL19, C9orf116, MRPS2, DCI, C10orf11, SCRIB, PBXIP1, PYGO2, C21orf84, CDK5, SLC4A2, NCRNA00114, STRN4, FKRP, CD300LD, KLC1, CNGB3, DARC, ARHGAP30, NGB, TSEN2, DDAH2, KLHL17, PLEKHN1, RYBP, GEM, FUT11, C9orf79, CRYAA, GP1BB, NOLC1, KRT15, FAM151A, HRCT1, LOC158376, KDM4B, PNMA3, ADAMTSL3, THAP11, TPRXL, TGIF2LY, TGIF2LX, GPR162, COASY, MLX, KIF2B, KRT6C, DCI, KLK6, RFNG, GPS1, C14orf79, ZNF683, PTGDS, LCNL1, SIGLEC7, GPR39, YDJC, CCDC116, SSRP1, P2RX3, FAM189B, LOC642587, FAM195A, WDR90, PCDH12, KIAA1967, ACTRT2, CALML4, TMEM179B, TMEM223, DDAH2, ROBO1, LRRC16B, SLC29A3, S100A3, KRTAP12-1, TMPRSS3, LOC90834, ST3GAL6, SIGLEC9, CDK3, PLA2R1, RFPL3, TLR9, XAF1, POU5F1B, CHPF, TMEM198, IP6K3, CRAT, PPP2R4, HDAC1, BTBD16, MEFV, SIGLEC7, DNAJC4, VEGFB, CRYBB3, CCR7, DCST2, DCST1, STXBP6, ABCB6, C2orf24, FAM134A, ATF6B, FKBPL, RAB37, TMPRSS13, UGT3A2, HAUS7, LY6G6E, LY6G6D, C9orf69, FAM65A, CCDC12, NBEAL2, CCL25, ASGR1, RASSF1, ZMYND10, EIF6, LOC643008, ENTPD3, MEFV, INPP5E, ST6GALNAC4, KRT4, NPW, LIMD1, SLC26A6, SLC2A7, LCN10, LCN6, MAP3K8, SLC25A45, CCDC28B, GRINA, PSAPL1, CTRB2, CSF1R, NMUR1, STOML1, PML, CA5A, PRDX2, RNASEH2A, MUC4, SCARF2, KRT6A, CARD9, C6orf126, C6orf127, LOC401431, ATP6V0E2, PDE11A, C21orf129, NCRNA00112, GPR81, NEK10, SMTN, STXBP6, FCGR2A, GRINA, PCK1, EDNRA, FAM118A, C9orf79, LOC400940, GRM4, CAMKV, DEFA1, DEFA1B, DEFA3, ATF6B, FKBPL, KRT1, XYLT2, LYPD4, DMRTC2, S100A13, S100A1, C9orf79, ATP2B4, PALMD, LOC26102, GPSM1, FAM151A, SCNN1A, CDH5, SIGLEC15, SOX15, BACH1, POLD1, RAB11FIP1, VTCN1, MYEOV, GRIA3, EREG, HSPG2, FGF11, CHRNB1, DENND2D, RUNX1T1, C14orf70, PTH2, CACNG1, PRICKLE2, MUPCDH, SCT, LGALS7B, PTMS, LAG3, C8orf74, CLDN4, GNGT2, ABI3, KCNE1, C8ORFK29, FBXL6, GPR172A, UBE2L6, UCN2, MAGEB6, ZNF19, GRIA3, GSDMD, GNAZ, LOC401431, ATP6V0E2, C19orf59, TRAPPC5, B4GALNT2, NXF4, MEIS3, ATP4A, C9orf116, MRPS2, ARHGAP27C10orf116, AGAP11, ACD, PARD6A, C16orf48, C16orf86, CPNE7, SH3KBP1, SCARF2, CD79B, SNAI3, LCN10, LCN6, DGCR6L, SH2D3A, VAV1, STBD1, ADAMTSL2, EP400NL, FPGS, WFIKKN2, KLHL5, FXYD3, PIP4K2A, SLC35A2, SMPX, EGLN3, MICALL1, ID2B, S100A9, FTSJ2, NUDT1, ARHGAP24, SHC1, CKS1B, CYP2J2, GANAB, INTS5, MRI1, MFSD10, C4orf10, NAT9, TMEM104, C14orf139, SHARPIN, MAF1, KIAA1875, C19orf59, TRAPPC5, RGS7BP, E2F2, KCNE1, TLR1, DVL2, PHF23, PLCXD2, IRF8, ADM2, MIOX, EPHA2, SSTR2, MYO1A, LZTS1, FLJ90757, BAIAP2, NEDD9, FBXW5, C8G, PNLIPRP2, SPEM1, RXRB, SLC39A7, HSD17B8, DHRS13, SERPINE1, C4orf50, MYH7, MUPCDH, SCT, UBXN6, C17orf61, NLGN2, TCP11, CANT1, FBXO6, SLAMF8, CXCR3, FBXW5, C8G, DYNLT1, SYTL3, HSPG2, MTA2, PARS2, LTA, TNF, C9orf103, LEPREL2, CCDC96, TADA2B, CXorf22, AARSD1, RUNDC1, SOX15, EPHB1, ATP2A1, FASN, PLA2G2E, KIAA1598, C9orf69, APPL2, SP100, FHL2, ITGB2, TRPT1, NUDT22, DNAJC4, MAP6D1, BTBD1, CXCL1, RFTN1, CCL25, GPR126, AOC2, ICT1, CCL17, POU5F1, PSORS1C3, OTOS, PARK2, PACRG, PRKAG3, C1orf56, VPS29, RAD9B, NLRP10, NDN, ANO7, NCLN, PRICKLE3, CTSZ, WFIKKN2, AK1, TMEM37, TUBB2A, KCTD11, TMEM95, KCNK18, CABP1, ANO4, RLN3, IL27RA, SSTR1, EIF6, BRMS1, B3GNT1, ERCC6, DHRS13, C2orf90, VWA3B, GUK1, LOC100240726, ENPP6, HOMER3, NLRP10, COMTD1, CIZ1, DNM1, IL25, CMTM5, CCR6, MARCKSL1, SHARPIN, MAF1, KIAA1875, IL11RA, C1orf231, MMACHC, KCNAB3, TRAPPC1, CNTROB, B3GNT5, ANGPT4, LOC100129637, DEFB124, REM1, PIGZ, FAM96B, CES2, ARMC3, ADRA1D, PTGS1, OR1L4, DAPK2, CMTM2, KCNAB3, TRAPPC1, CNTROB, RAB4A, PRKCH, GPR126, BBC3, CUEDC1, PHLDB1, MAGEB6, HABP4, DLGAP2, TLR2 | Figueroa et al., 201352 |
| S100A16, HNRPLL, DNAJB2, SH3BP5, LARS2, RGS12, RBPJ, DDIT4L, ANKRD33B,RP1-209A6.1, TNXB, CLEC5A, PKIA, EIF2C2, LCN6, C9orf139;FUT7, RP11-393K12.2, MARCH8, ALDH3B2, RAD51L1, SYNE3, SEZ6, MOBKL2A, PLVAP, CLDN14, LOC284837, CELSR1 | Nordlund et al., 201553 |
Methylation profile of other subtypes of pediatric ALL-B.
| Subtype | Methylation profile | Reference |
|---|---|---|
| Translocated MLL, BCR-ABL and TCF3-PBX1 | Hypermethylated genes:WWC1, NELL1, SEMA3C, FGFR2, ARNT2, NRG1, FBN2, KCNC3, SSH3, DSC3, CYB5R2 | Chatterton et al., 201451 |
| Hypomethylated genes:CHST12, LSP1 | ||
| Translocated MLL | Hypermethylated genes:ZAP70, XYLT2, HLA-B, EDEM1, UBXN11, CD52, C14orf43, HLA-C, MICALL1, C14orf43, PRKCH, TP53I11, GHRL, SNORD10, SNORA67, CD68, MPDU1, ITIH3, CMTM2, PTPRE, MOV10, GUK1, LOC100128164, SEC62, IFITM2, SELO, TOR2A, C3orf37, CD79B, VWA5B2, CD74, C22orf26, LOC150381, ARF1, SORBS3, APPL2, AK1, CCDC102A, GPR114, KPTN, PPP1R14B, PLCB3, KCNK3, LRRC15, ITPRIP, GAB1, PPP1R14B, PLCB3, GP9, CACNB3, FOXN3, FDX1L, AFF3, SNORA77, PRX, CLINT1, UCKL1, UCKL1AS, CAMTA2, FTSJ2, NUDT1, TMEM220, DLGAP2, AMIGO3, GMPPB, KPTN, ANKRD13D, NINJ1, SSTR2, PPP1CA, TBC1D10C, SIK1, SBSN, GAPDHS, PRKCB, IL2RA, KLHDC7A, DEGS1, LIMD1, AK1, CTCF, ZNF267, CCR6, ANGPT4, DENND3, LTB, LST1, MDC1, TUBB, FLJ42393, MCF2L, C1R, HPCAL1, C21orf84, SPAG8, HINT2, SLC19A1, PLA2G4D, NLRP12, GUK1, PRSS27, LSM7, SPPL2B, GADD45B, TMEM220, TMEM204, ANKRD13D, CTDSP2, SH2D4B, STK32C, EFNA3, ANGPT4, SPIB, IRAK3, NDUFB11, RBM10, APP, ARHGAP27, TMEM129, TACC3, SMTN, SPAG8, HINT2, DNAJC10, S100A13, S100A1, NUP107, HIRA, MRPL40, TENC1, OAF, C7orf61, DBNDD1, RPS11, SNORD35B, HSPA8, TAP1, PSMB9, RPAP1, REPIN1, NUDT8, DNTT, STRN4, FKRP, LTA, TNF, TMCO4, HABP4, NCK2, SSTR2, USP50, CXCR5, GYPC, XYLT2, B3GAT3, SIRT2, ARHGAP24, S100A13, S100A1, BCL7C, CTF1, COG8, NIP7, PTPN18, ZFP161,SLC27A2, PPP1R3E, BCL2L2, ITIH3, LOC401431, ATP6V0E2, STK32C, RAB37, MYO7A, GUK1, GJC2, PCNA, CDS2, STK38, HSPB11, LRRC42, STAB1, GP9, NCLN, RGS14, C21orf84, SERPINE1, COG3, CPNE7, BTK, RPL36A, SGPL1, MRPS15, PIK3CG, CRAT, PPP2R4, MDM4, IL3, MLL2, C1orf231, MMACHC, LOC401431, ATP6V0E2, CD22, FFAR1, MAP6D1, BCL7C, CTF1, TMEM127, CIAO1, CNR2, PXK, STX17, ADCK4, ITPKC, CPNE7, CD79B, SHARPIN, MAF1, KIAA1875, IL3, PELI3, SIGLEC15, C3orf42, GHRLOS, NUP205, MAP6D1, PRDM8, DHRS13, FASN, MUPCDH, SCT, LOC100127888, GRINA, RFTN1, IP6K1, AKAP8L, NCRNA00095, FBXL19, MTHFR, CLCN6, C22orf26, LOC150381, PIP4K2A, LOC100128164, SEC62, DAXX, CYFIP2, TNF, LTB, GANAB, INTS5, CLDND2, NKG7, CD37, C12orf49,RNFT2, POU2AF1, SOX15, ZNF366, TMEM127, CIAO1, SFN, LOC100127888, KRTAP5-8, SUV39H2, FAM189B, TIMM17B, PQBP1, ANKS6, TERT, KDM4B, C9orf116, MRPS2, GRINA, ZNRF3, POMP, MAG, MYL4, RHOH, DNAJC10, UGT3A2, BTN3A2, C22orf24, YWHAH, ZNF566, TINAG, ZFHX3, PBXIP1, TGM6, S100A13, S100A1, ACTRT2, ALPPL2, WIPF1, RECQL4, LRRC14, NCKIPSD, R3HCC1, RAB24, PRELID1, SOX15, LY6G6E, LY6G6D, HPS4, SRRD, CANT1, ADM2, MIOX, ATP2B3, SNORA67, CD68, MPDU1, FLJ25006, LOC645851, C21orf129, NCRNA00112, LEPREL2, LTB, LST1, GPER, DHRS13, MUC4, COG3, GNB4, RANBP9, DCUN1D2, TMCO3, RAB33B, ST6GALNAC3, C1QTNF4, STBD1, POLD1, LRRC6, GFAP, LRFN2, MTMR10, FITM1, PSME1, SFT2D1, CBLN3, KHNYN, DAPK3, HBEGF, SIDT1, FUCA1, CSDA, C14orf49, VKORC1, CISH, MAPKAPK3, RAB11FIP1, GFAP, KRTAP12-1, ITSN2, CIZ1, DNM1, TMED6, MAN2A2, MAP1LC3A, SORL1, RAG2, C11orf74, TIMM13, KLHDC5 | Figueroa et al., 201352 |
| CDH3, TBX2, ERCC1, NPR2, DAPK1, CCR6, HRK, LIFR1, DLX3, FHIT | Stumpel et al., 200960; Schafer et al., 201061 | |
| Hypomethylated genes:GUCY1A3, JMJD1C, LOC84989, BAZ2B, PRF1, RUNX3, PRF1, MAP7, RHOBTB3, RUNX3, LARGE, SLC25A18, ITGAE, FLT3, NPR1, FAM65B, PTGR1, ACSL1, MEF2C, TRPV3, SKA1, CNKSR2, H1F0, GCAT, F3, MTHFD2L, TLR7, ARRDC4, DAPK1, PSMB5, TMEM41B, LOC100303728, SLC25A5, ZNF367, FLT3, LOH12CR2, LOH12CR1, PHLPP1, SERPINB13, OR6V1, PRDM11, CD3D, CD3G, SENP6, TRAK2, STRADB, ASTN2, TRIM32, SETBP1, IGF2BP2, TFCP2L1, CENPF, MPP5, C7orf13, RNF32, PSMB5, C7orf13, RNF32, CD180, PARP8, RBKS, LOC100302650, BRE, ARID1B, C6orf122, C4orf19, MGC16703, P2RX6, OLFML3, IQSEC1, MYST2, KRTAP10-6, PDPN, NID1, CPD, LCE3A, SSR1, CDKL3, UBE2B, PAPOLA, ANKRD27, RGS9BP, HTR7, DES, FAM5B, CDC14B, GPR153, SPOCK2, ITGAE, LECT2, SERPINB8, ZAR1L, BRCA2, CAMTA1, C9orf100, ALDH4A1, C17orf64, KRT81, LILRB4, MRPS25, ASB8, SLC25A38, TRAS, MYOD1, DKK3, PPP1R3B, BDNF, CCBL1, LRRC8A, RASGRF2, GRIK5, KCNJ10, FAIM, MTMR11, PEA15, SBF2, HECW1, WDYHV1, PHLDB2, GINS3, PDE4DIP, KDM5B, ZNF256, PHLDB1 | Figueroa et al., 201352 | |
| ZSCAN18, ZNF256, ZNF329, ZNF544, ZNF681 | Nordlund et al., 201353 | |
| BCR-ABL | Hypermethylated genes:KIAA1949, NRM, MUPCDH, SCT, ATP2A1, FASN, DPM2, PGLS, MYEOV, DUSP5, MARCKSL1, ANKRD27, RGS9BP, ADCK5, FOXH1, C11orf16, DIRAS1, MRPL24, DCST2, DCST1, CCDC71, INPP5E, SLC22A20, GPR146, PLEKHM2, PROC, DIRAS1, ALDH4A1, WFIKKN2, IL11, LIMS2, GPR17, GRINA, LILRB4, C19orf59, TRAPPC5, CD38, LOC100129637, PRPH, LOXL4, KLHL24, ARHGAP27, GPR146, SSR1, C19orf59, TRAPPC5, C17orf61, NLGN2, C9orf116, MRPS2, DLG4, ACADVL, TLR9, STOML1, PML, KLF13, KCNK17, KLC1, ARHGAP30, C8ORFK29, FBXL6, GPR172A, DLG4, ACADVL, TAF6, CNPY4, MBLAC1, C9orf69, AURKC, PCOLCE, PTMS, LAG3, DNM1P35, GRINA, BSCL2, GNG3, CCL25, TEX19, CDH5, TLR9, CDK5, SLC4A2, GUK1, C9orf116, MRPS2, S100A13, S100A1, FXYD2, GPR153, LOC100129637, FLJ90757, BAIAP2, IP6K3, KLF6, LPAR2, UCN2, KCTD11,TMEM95, TFR2, C14orf70, CARHSP1, KLF13, CCDC96, TADA2B, LIMS2, GPR17, COL1A1, ARID3A, SHARPIN,MAF1, KIAA1875, SIRT2, WFIKKN2, ZNF296, GEMIN7, RFNG, GPS1, SLC26A8, MAPK14, FAM176B, CCBL1, LRRC8A, KCNK17, KCNAB3, TRAPPC1, CNTROB, MAGIX, PCDH12, SHARPIN, MAF1, KIAA1875, C16orf59, C9orf69, SP100, LACE1, C17orf64, CSF1R, MUC4, C1QL4, KCNAB3, TRAPPC1, CNTROB, GRB7, CLDN4, CARD9, ATP1A3, C1orf220, GUK1, FAM109B, C22orf32, CLTB, HSF2BP, RRP1B, GABBR1, B3GNT5, ITIH3, LOC26102, GPSM1, UBXN6, SH3D20, SIGLEC7, EDC4, CCR7, ACTRT2, CTXN1, RAB3IL1, DNAJC4, VEGFB, ADM2, MIOX, TUBB2A, SSX2IP, C16orf81, LIMS2, GPR17, ZNF48, PKNOX2, GRASP, PECR, TMEM169, NTN5, C9orf103, CACNA1I, BTG2, ADRA1D, DHRS13, THEM4, MT1L, PHLDB2, PDE4DIP, IL25, CMTM5, LIF, KLHDC5, PITPNM3, GALP, FAM109B, C22orf32, PIGZ, PRDM11, CNTN2, MIB1, BSN, MOCOS, DRD5, COL1A2, KIAA1257, LYPD4, DMRTC2, POU5F1, RASL11B, GFAP | Figueroa et al., 201352 |
| Hypomethylated genes:GBA2, RGP1, DPP3, C11orf30, ECHDC1, ZEB2, INHBE, GLI1, INTU, WBP11, C12orf60, STARD3NL, C21orf59, FGF6, ITGA2, FBXW7, OIP5, NUSAP1, C6orf47, BAT4, CSNK2B, RPS13, RPL4, SNORD18C, SNORD18B, SNORD16, SNORD18A, ZWILCH, CCDC115, IMP4, POLR1E, C22orf26, LOC150381, ANG, RNASE4, SRBD1, GIMAP5, C1orf183, SLC25A28, TRIB3, CTNNA2, STBD1, C22orf26, LOC150381, IQGAP2, RHBDD3, EWSR1, CSDA, C3orf42, GHRLOS, CYP1B1, GPR6, MFI2, TTC23L, OGFRL1, LRRTM1, CCDC70, C5orf43, NKX2-1, ADAMTS12, NDUFC1, NARG1, TUBA4A, TUBA4B, CLIC2, TMEFF2, KIAA0087, UGGT2, CACNA1B, PRPF8, PJA2, CXCL12, SLC20A1, ACSBG1, CA6, PDZRN3, RTDR1, RAB36, ROBO4, BRDT, PROX1, COL14A1, FLJ14107, ATOH1, NLGN4X, SLC35A1, LRIG3, LANCL1, CPS1, HOXD1, KCNN2, PCNA, CDS2, IL2RA, ZNF264, PCDHGB7, PCDHGA11, GAB1, FGF5, SALL3, CISH, MAPKAPK3, C15orf24, PGBD4, SFN, RIC3, FAM190A, LIMD1, MTMR11, EPHA5, IRX2,C5orf38, ZNF804A, HIPK2, TBXAS1, TINAG, BPNT1, IARS2, MSX2, GLIPR1L2, PRDM8, BDNF, MTERFD2, HOXD1, LRFN5, CDCA5, ZFPL1, MAL2, CDH8, CD22, PCDHB18, PCDHB19P, KCNK13, PKD2L2, GZF1, PDPN, VKORC1, DAXX, FOXQ1, TNFRSF9, MRPL1, TRIM13, UCHL5, TROVE2, EFR3A, SLC39A1, CREB3L4, WIPF1, XYLT2, CPVL, RAG2, C11orf74, PCDHB7, PCDHB8, BCL2L13, METTL8, DCAF17, LRP11, CHMP4C | Figueroa et al., 201352 | |
| TCF3-PBX1 | Hypermethylated genes:FNBP4, CLDND2, NKG7, ARPP-21, SOCS2, NCK2, ARHGEF6, GNA13, TLR1, TMEM156, CD79B, HABP4, ISG20, FOXM1, C12orf32, LAIR1, HECA, CST4, STX17, NFKBIE, TMEM151B, PIGV, MLKL, FDXACB1, C11orf1, ASGR2, EFHC2, GNGT2, ABI3, USHBP1, C19orf62, ZNF25, OAS3, CYSLTR1, GAB1, GNGT2, ABI3, SORBS3, KRAS, BEST1, ARAP3, POLD1, HSPB11, LRRC42, RAB37, ITPRIP, MTA3, KIAA2013, SERINC5, ARHGAP24, KLHL5, CCL25, IL2RA, B3GNT9, TRADD, FBXL8, UBE2L6, KLHL21, GABBR1, OLFML3, NGF, C5orf56, PCNA, CDS2, SSRP1, P2RX3, TMEM127, CIAO1, STAB1, ETFA, TSFM, LOC100129066, ZNF683, ARHGAP24, HSD17B4, CPNE8, NUP107, UGT3A2, FBXO6, CLEC4D, VRK2, C11orf75, PDK1, CCL17, PSTPIP1, CEP350, IKBIP, APAF1, VWA3B, HGF, TMEM217, TBC1D22B, C17orf64, RNF113A, NDUFA1, HELZ, BTG2, SMARCB1, EIF4E3, GPR27, MEF2C, DUSP4, CTDSP2, NUDT5, CDC123, ATP5G2, ST8SIA1, PRDM8, CTCF, SMTN, CMTM2, DAXX, KLHL24, CACNA1I, NCLN, PTPLAD2, ERCC6, HBEGF, PRDX1, FCGR2A, CLU, TNFAIP8, LTB, LST1, FOXN3, MCM3AP, C21orf57, SMC1B, RIBC2, BTG2, SMC6, GEN1, DAZAP2, SYNGR1, CDC2L5, LMAN2, UTP6, C22orf24, YWHAH, SIGLEC15, LOC646999, STRA6, NOLC1, NCAPH, SENP6, ZNF423, NCKIPSD, PPP1R15B, SERPINE1, KPTN, FAM60A, FLJ13224, LDLRAD3, LCN10, LCN6, SLC38A5, FTSJ1, SPOCD1, FPGS, ANKS6, WIPF1, MTMR11, MRPS15, NOLC1, C20orf197, ZNF652, DPY19L3, ATP2B4, SRGAP3, EN1, ZNF41, CYBRD1, RANBP9, LRP11, LTB, LST1 | Figueroa et al., 201352 |
| Hypomethylated genes:ZNF512, CRYM, WAPAL, SLAMF1, MYBPH, EXTL3, FAM109B, C22orf32, LHCGR, AKR1B1, SURF1, SURF2, PLXNC1, WDR87, SIPA1L3, NOL11, EXTL3, SDF4, B3GALT6, DEF6, NCRNA00176, LIPC, NFKB1, ASB8, TERT, NSUN4, LOC100129534, ADIPOQ, PHYHD1, BLK, LPAR2, FAM109B, C22orf32, GJA4, ADIPOQ, PDCD1, MND1, RASL11B, RNF186, PSMC3IP, MRPL28, ACTL9, TNS4, LAT, FAM109B, C22orf32, TEKT2, ADPRHL2, NAGA, FAM109B, C22orf32, PLD4, TEKT2, ADPRHL2, C16orf11, GLYCTK, NGF, PIP4K2B, ACOXL, MAST4, AURKC, SOST, MAP6D1, POLM, MSRB3, VAMP2, SLC39A1, CREB3L4, IDH2, NAT1, SSR1, ST6GAL2, FXYD2, ST6GALNAC3, CDCA3, USP5, PHACTR3, FFAR1, SDF4, B3GALT6, GDPD5, ARL4C, ATXN7L2, FAT1, GLYCTK, TRIM67, CTBP1, C4orf42, ARL4C, HOXD1, TSPAN3, TBX6, YPEL3, B3GNT7, MUPCDH, SCT, HIST1H1C, ITIH3, C12orf53, SOX21, SALL3, HECW1, PEA15, MNAT1, WNT2, PPARD, C16orf81, LILRB4, WNT2, C9orf100, SOX9, NCRNA00176, TFAP2C, C16orf81, GPR123, HOXD1, NXPH2, USP11, LTA, TNF, SCAP | Figueroa et al., 201352 | |
| Rearranged CRLF2 | Hypermethylated genes:STXBP5, SLC2A8, GFRA1, DSC3, C9orf150, LCE3A, CHRDL1, C15orf24, PGBD4, HOXB2, ST6GAL2, NOVA1, NOL4, SLITRK1, TMSB15A, PRPF19, LRRC57, HAUS2, C18orf34, EPHA5, DSC2, RBPMS, TUBGCP3, AQP4, C18orf16, GABRQ, TMTC1, AADAT, ZIC5, MYEOV, ASXL1, NEUROG1, EPHA5, SIX6, TMED3 | Figueroa et al., 201352 |
| Hypomethylated genes:HIPO, LYSMD4, SRBD1, PYGM, CASQ1, KCTD3, ANO7, WIPF1, CA6, GP1BB, NLRX1, LCN10, LCN6, SLC2A5, S100A13, S100A1 | ||
| Altered ERG | Hypermethylated genes:CDKN2C, CYSLTR1, TMEM156, PIGV, RFTN1, SOCS2, MTHFS, MLKL, SERINC5,CD48, FLJ14107, ENAH, TNF, LTB, DAXX, RFTN1, TNFAIP8, TUBA4A, TUBA4B, HIBCH, NUBPL, C9orf25, DNAI1, STIM2, CHMP1B, PEX16, GYLTL1B, TLR1, CH25H, DEGS1, C8orf56, BAALC, XPO5, POLH, CXCR3, MED14, DDX3X, DENND3, ZNRF3, PELI3, PIK3IP1, HELZ, TGM6, GAB1, RBP1, C1QL4, ATP2B4, PRR5L, SLC27A2, ATP10A, LTB, LST1, STIM2, TMEM178, BNIP3L, TNFAIP8, CEP350, ZMYND8, UBE2J1, CLINT1, EXOSC9, MTMR10, FPGS, CCNI, CDKN1A, UBXN11, FAM120AOS,FAM120A, SYNGR1, ITGB2, RNF141, CD3D, CD3G, SRGAP3, CHRNA2, ZNF490, ZNF791, TP53BP1, WBP11,C12orf60, WDFY3, C4orf12, CARD8, GGNBP2, IKBKE, UTP20, RAPGEF1, HSPA8, PPP1R15B, SENP6, ST6GALNAC3, CD48, KDM4B, PPID, ERO1LB, SH2D3A, VAV1, KCTD17, ZNF212, ARHGAP24, RANBP10, TSNAXIP1, CYTIP, LTB, LST1, LOC729234, PKNOX2, C8orf56, BAALC, SYNE1, DYNLT1, SYTL3, TRIM13, UBR5, IGFBP7, ERAP2, CDCA5, ZFPL1, PARS2, MEF2C, PIP4K2A, NLRP12, ANKFY1, ST3GAL6, PJA2, UGT3A2, DHRS13, FUCA1, CENPJ, RGMA, RAB39, LRRC8D, RANBP9, AIF1, ITPRIP, TAF3, ERCC6, MRPS15, SRGAP3, CDCA3, USP5, NGRN, PIP4K2A, L1CAM, SERPINE1, EFHD2, FXYD5, NUP107, NARG1L, MAP3K8, C14orf33, KTN1, RPRML, RNF113A, NDUFA1, ADAMTS5, MDM4, TLR7, EGLN3, HYAL3, NAT6, HYAL1, NOVA1, DCTN2, KIF5A, TRIM9, ST6GALNAC1, C1orf127, WIPF1, LRRC57, HAUS2, CYBRD1, SLAMF1, ACOXL, PBK, ECM1, RASL11B, TMEM37, GRPEL2, ACSL3, ICAM3, C1orf156, C1orf112, PXT1, KCTD20, STXBP6, TBCCD1, DNAJB11, SIDT1, FGFR4, SLC16A9, CAB39L, SETDB2, ERICH1, DTX3, SSTR2, CBX6, VRK2, C14orf48, EIF4E3, GPR27, SMCHD1, XYLT2, TSEN2, CEP70, STXBP6, USHBP1, C19orf62, ST6GALNAC2, ADAP2, GLRA2, WFDC2, NOLC1, GFI1, CD180, NP, SLC39A13, IRAK3, SPRYD5, C9orf79, DNAJC9, MRPS16, LYPD5, ZNF283, TMEM217, TBC1D22B, ENTPD3, HSPG2, CCDC85A, STK38, CNR2, KIR3DX1, IL8RA, CECR2, MYL12A, MYST2, KCNAB1, WDR47, PYY, NAGS, GNAZ, DEFA1, DEFA1B, DEFA3, C17orf64, RPL17, SNORD58C, U58, SNORD58A, SNORD58B, ESPL1, PXK, CARD14, HNF4A, C9orf79, PCDHB2, CD33, DGKG, S100A13, S100A1, FIBIN, ATP5G2, RAB11FIP1, GREB1L, GJA4, PRICKLE2, RAG2, C11orf74, ZCCHC9, ZNF683, CXXC5, HNMT, C12orf23, ACSBG1, CLU, BPNT1, IARS2, CCR7, ALX3, GABRR1, TFB2M, C1orf71, DDX19A, CACNA1S, THYN1, ACAD8, PTH2, KIF12, GNGT2, ABI3, CAMK2B, SEMA4A, ADARB2, MC4R, DNAJC10, TMPRSS13, C7orf43, DOCK3, DCK, NBLA00301, STBD1, USP2, CDR2, SSRP1,P2RX3, IKBIP, APAF1, PNPT1, DCAF5, ZNF423, PLEKHH3, CCR10, CNTNAP1, LEMD1, PCDHB7, PCDHB8, MLL2, XPNPEP2, DYNC2H1, CPOX, PIAS2, GMNN, HMP19, SEL1L3, PTPRB, CRYBB3, C14orf115, NELL1, ITIH5, RRAGA, LRAT, DEFB124, REM1, C15orf24, PGBD4, B4GALT3, RHBDL3, SRBD1, HTR7, FAM193B, GNB1L, C22orf29, AFAP1, BRDT, MRPL1, DIAPH2, PRICKLE3, ACTRT1, TNFRSF9, SSTR2, HNF4A, SOCS3, SLC39A1, CREB3L4, KCNAB3, TRAPPC1, CNTROB, FOXO4, NOV, NOLC1, DNAJB1, C9orf79, LOC400940, TMPRSS2, GPR84, ZNF366, SFT2D3, TUSC3, VPS72, DCBLD1, AMMECR1L, SLC2A14, PRR18, VPS24, DNAJC10, EXOC8, C1orf124, MAL2, TPRXL, YARS, S100PBP, PO | |
| Hypomethylated genes:A2LD1, SAP18, BTN2A1, OSBPL11, HN1, AARS2, ADI1, OBFC2B, SLC39A5, ZNF295, PARVB, MIA, RAB4B, ZNHIT2, FAU, MRPL49, TULP4, C7orf65, TMIE, PSMA2, MRPL32, SPINK4, ZNF296, GEMIN7, PEX11A, WDR93, RASSF2, PNRC1, SLC39A11, GIT2, ANKRD13A, DIXDC1, VPS4B, PARVB, CHRNA1, KRTAP5-8, ADAMTS9, CNTN2, C14orf139, EPS8, PCNX, LDOC1L, CCDC109A, PRMT10, CCNT1, NSD1, WDFY4, DCP1B, GCHFR, C12orf36, RFC4, TFF3, GPR81, ZNHIT6, C19orf23, C19orf24, PIK3R1, C16orf11, NHLRC4, PIGQ, C10orf118, TDRD1, XPNPEP2, SECISBP2, GLYCTK, CUL7, MRPL2, KLC4, GLG1, C7orf25, GPR153, EIF1AD, BANF1, PPL, TMEM127, CIAO1, GPR146, CAND2, DIXDC1, UCKL1, UCKL1AS, SNHG7, SNORA43, SNORA17, RFC4, NME3, MRPS34, EME2, ING1, USP12, HNRNPR, RGN, NHLRC4, PIGQ, TSKU, GRB2, CST8, SCGB3A1, ZNF513, ADCY1, LRRN1, C3orf27, EXOSC10, LFNG, XYLT1, C14orf126, GPR44, CRBN, C16orf81, GLIPR1L2, LTA4H, SPIB, CCNJ, GPR146, LOC643008, GLIPR1L2, BACH2, CCDC109A, GPR155, ZC3H18, ELP3, ALPPL2, CRBN, ADAMTS3, POLM, MRPL28, HEATR5B, CCDC75, GPER, CUL7, MRPL2, KLC4, CHRNA1, C14orf139, SREBF1, MRPL55, KCNC3, BTG2, MTSS1L, OR2Z1, LOC100303728, SLC25A5, CACNA1I, IFI30, GRINA, CRHBP, RNASE7, CD81, SURF1, SURF2, PTOV1, TGFBR2, TEKT2, ADPRHL2, ADAMTS3, IGSF21, SOHLH1, KCNT1, RPL9, LIAS, SHARPIN, MAF1, KIAA1875, GRINA, C20orf165, NEURL2, CTSA, SLC20A2, C8orf40, SNW1, C14orf178, RHBDF1, MPG, LOC26102, GPSM1, EIF4G2, RHBDF1, MPG, TPI1, GLYCTK, ATP2A3, ARHGEF4, TBC1D13, CD38, MS4A10, LOC100129637, SOX15, LIF, C19orf23, CIRBP, C19orf24, ABCA2, C9orf139, CCDC96, TADA2B, BSCL2, GNG3, NELL1, RGS9, GPR148, GANAB, INTS5, HIST1H1C, BMP7, LOC100129637, CGREF1, ABHD1, PCP2, STXBP2, LOC643008, CIZ1, DNM1, GPER, TSKU, TRIM67, GFRA1, FLJ90757, BAIAP2, FTSJ2, NUDT1, DDX50, FOXJ2, SLCO3A1, LIN7A, GATM, RAMP1, COG8, NIP7, NFS1, ROMO1, VPS37C, FAM53B, G0S2, SHANK1, SPATA24, C19orf23, CIRBP, ZNF566, FAM83H, IMMP2L, LRRC3, TSTA3, HLF, SDF4, B3GALT6, LHCGR, C16orf42, GNPTG, SLC2A8, HS6ST1, C20orf165, NEURL2, CTSA, SFT2D1, CSMD1, RAD54L, PITRM1, MUPCDH, SCT, TIGD3, VAT1L, TOMM40, CCDC42, WARS, WDR25, GRIK5, CYP2U1, ODF3L2, HMP19, KLHL22, ARID3B, MT3, SOX15, DMRT3, REPIN1, MTA2, ATPAF2, C17orf39, SHARPIN, MAF1, KIAA1875, SQLE, CHMP4C, SYT15, MYH6, SNTB1, MRPL24, GPR123, GATA6, FBXW5, C8G, EPHA5, PRELID2, IVNS1ABP, KRT80, SNAI2, RBM3, VWDE, NUDT10, ZMYM6, MRPS30, TIMM13, ZNF532, SYVN1, NKX2-8, KIAA1024, FAM81A, KCNN2, MUC1, THYN1, ACAD8, LOC100130557, NFYC, ALOX5, CCDC58, FAM162A |
Figueroa et al. evaluated patients with ALL with both immunophenotypes. In their study, included 137 children with ALL-B and 30 with ALL-T and identified a signature of 85 genes with alterations in methylation, which correlated with the expression analysis (Table 2). The identified signature was common between all ALL subtypes (B and T) and included genes TIE1, MOS, CAMLG, GPRC5C, which are involved in signaling; MCTS1 and DGKG, which participate in regulation of the cell cycle and cell proliferation; PABPN1 and PABPC5, which participate in RNA metabolism; PROP1, TAF3, H2AFY2, ELF5, ZBTB16, CNOT1 and TADA2A, involved in transcriptional regulation, as well as homeotic genes HOXA5 and HOXA6.52 Additionally, this study determined specific methylation and expression signatures characteristic to every ALL-B subtype in children. Seven independent ALL-B groups that presented a characteristic methylation/expression signature, depending on the molecular rearrangement present in each group of patients were identified: rearranged CRFL2, ETV6-RUNX1, high hyperdiploidy, translocated MLL, altered ERG, BCR-ABL1 and TCF3-PBX1 (Tables 3-5).
The study by Norlund et al., which included a greater number of patients with pediatric ALL (663 children with ALL-B and 101 children with ALL-T), confirmed that each ALL subtype presents with a specific methylation signature. Based on this study, it was proposed that methylation signatures could be useful to predict the genetic subtype of pediatric ALL. This proposal would be applicable to cases in which difficulties arise with the identification of a chromosomal or molecular alteration that contributes to leukemia classification.53 Despite methylation signatures to determine ALL subtype have been found and epigenetic profiles to allow the prediction of the risk of relapse have been proposed,47,54 an epigenetic profile capable of accurately classifying and stratifying patients with ALL does not exist.48
3.2Alteration in gene methylation in ALL according to the specific subtype of genetic rearrangementBesides alterations in DNA methylation, which are common to all subtypes of ALL-B, exclusive modifications to each genetic subtype exist.48,51,55 Recently, methylation profiles together with gene expression profiles in patients with the ETV6-RUNX1 fusion were analyzed; 55 genes with alterations in methylation were identified (Table 3).51 Among these alterations, EPOR gene was identified, which was found to be overexpressed, and its promoter, hypomethylated. The ETV6-RUNX1 fusion binds and activates the transcription of EPOR (erythropoietin receptor), which contributes to leukemia development, activating cellular proliferation and survival through the JAK2-STAT5 pathway.56 Hypomethylation in EPOR favors its promoter to be more permissive to binding and activation by ETV6-RUNX1.
In the ETV6-RUNX1 subtype, hypermethylation and low expression in the asparaginase synthetase (ASNS), which sensitizes cells to treatment with L-asparaginase was also found. This explains, in part, the fact that patients with ETV6-RUNX1 present such a good treatment response.57,58 Other genes with alteration in methylation and expression observed in ETV6-RUNX1-positive patients include CLIC5, ACVR1C, IGF2BP1, DSC2, PCLO, SOX11, SPSB1, BEST3, SIDT1, TCFL5, CHL1 and FAM19A (Table 3).55,59 Additionally, it has been reported that alterations in the methylation of ERHV-3, DMNBP, KCNA3, PAG1 and C11orf52 are associated with risk of relapse in patients with ETV6-RUNX1, for which they have been proposed as markers of poor prognosis.47
In patients with hyperdiploidy, alterations in methylation and gene expression that are exclusive to this subtype have been found. More recent studies highlight the hypermethylation and low expression in tumor suppressor genes FHIT, PTPRG and DDIT4L.47,51 Additionally, hypomethylation of proaptotic genes BCL2 and BCL9L has been observed, which have been reported to be altered in diverse B cell tumors (Table 4).51 On the other hand, the ALL subtype positive to rearrangements in the MLL gene also presents a characteristic profile of alteration in methylation (Table 5). It has been reported that in ALL that with rearrangement in MLL, hypermethylation of CDH3, TBX2, ERCC1, NPR2, DAPK1, CCR6, HRK, LIFR1, DLX3 and FHIT genes occurs.60,61 Hypermethylation of genes ZSCAN18, ZNF256, ZNF329, ZNF544 and ZNF681 has been associated with an increase in the risk of relapse in patients with alterations in the MLL gene.47 In particular, it has been observed that a different methylation profile is present in ALL with different translocations in MLL.60
3.3Main cellular processes and signaling pathways affected by alterations in gene methylation in ALLCell processes with a greater representation of genes with aberrant methylation in ALL include the regulation of the cell cycle, apoptosis, transcriptional regulation and cellular adhesion (Figure 3). Some examples of genes with alterations in methylation in ALL that participate in these cellular functions are CDKN1A, CDKN1C, CDKN2A, CDKN2B, SFRP, CDK6, which regulate cell proliferation and cell cycle62–66; GATA4, HLF, TCF3 FOXF2, PAX2, PAX5, PAX6, TFAP2A, TWIST72, TP73, HOXA4, HOXA5 and HOXA6, which participate in gene transcription 51,62,67–70; DAPK1, APC, EPHA2, RASSF1, PTEN, APAF1, DBC1, DCC, EPHA2, EYA2, miRNA34, miRNA34C, NOXA, ASPP1, PTPN6, RASSF6, SFRP1, WDR35, PAR4, ERBB4, involved in apoptosis4,20,36,51,71–73. Alterations in the methylation of these genes confer leukemic cells with survival advantages, preventing apoptosis and promoting proliferation (Figure 4).


Some signaling pathways mostly represented by genes with alterations in methylation in ALL are p53, WNT, EPHR, MAPK, and PI3K-AKT4,20,59, which are tightly related to cell processes as the aforementioned (Figure 4). One of the most profoundly studied pathway is the TP53, which is a tumor suppressor gene involved in the activation of damaged DNA repair, induction in the G1/S checkpoint and promotion of apoptosis.74 Despite mutations in this gene are present in 50% of tumors, less than 3% is found in ALL. However, an alteration of the p53 pathway at the epigenetic level exists in ALL.59 Hypermethylation of at least one gene involved in the p53 pathway has been observed in 78% of patients with ALL. Hypermethylation in genes involved in apoptosis dependent on p53, such as AIFM2, APAF1, DBC1, miRNA34B, miRNA34C, PMAIP1, POU4F2, PPP1R13B, TP73, NOXA, AMID and ASPP1 has also been detected. In the same way, hypermethylation of genes such as CDKN1A, CDKN1C, CDKN2A, POU4F1, which participate in cell cycle control dependent on p53, and hypermethylation of LATS2 and DAPK1, which participate in the regulation of this same pathway.4,20,59,71,75
The WNT/β-catenin pathway has been involved amply in diverse types of cancer.76 Activation of this pathway regulates proliferation and cell differentiation, among other processes.4 In ALL, an alteration regarding methylation of genes associated with the WNT, such as WNT5A, RSPO1 and APC has been reported.36,72,77 Additionally, aberrant methylation of sFRP2, sFRP4, sFRP5, WIF1, DKK3, sFRP1, PTPRO, FZ10 and DKK2 genes, as well as in β-catenin inhibitor, cadherins (CDH1, CDH11, CD13), and genes of the SOX family (SOX2, 3, 8, 9, 11, 14, 21) has been reported in patients with ALL-B with relapse.54
Other signaling pathways affected in ALL by alterations in DNA methylation include the ephrin receptors (EPH), which are tyrosine-kinase receptors that activate and regulate diverse biological processes. In ALL, it has been suggested that EPH genes can act as tumor suppressors. Hypermethylation in receptors and ligands, including EPHA2, EPHA4, EPHA5, EPHA6, EPHA7, EPHA10, EPHB1, EPHB2, EPHB3, EPHB4, EFNA1, EFNA3, EFNA5, and EFNAB1 has been reported.36 Additionally, alterations in methylation of the MAP kinase pathway genes (MAP2K1, BRAF, ERK2) have been reported in ALL, as well as in the PI3K-AKT pathway.54
4Histones modification in ALLHistones are proteins involved in DNA organization inside the nucleus. There are five types of histones: H1/H5, H2A, H2B, H3 y H4. Histones H2A, H2B, H3 and H4 form the nucleosomes which pack the DNA, whereas histone H1 is found in the spaces between the nucleosomes; histone H5 is present in specific regions of the DNA. The state of the chromatin depends on post-translational modifications in histones and influences gene transcriptional states. Numerous covalent histone modifications are known, such as acetylation, methylation and phosphorylation, which lead to gene repression or activation (Figure 1).4,9 The predominant and most studied modifications are lysine and lysine/arginine methylation.9
The main enzymes in charge of regulating histone modification are the methyltransferases (HMTs), demethylases (HDMs), acetyltransferase (HATs) and deacetylases (HDACs).78 The combination of the activity of these enzymes confers a “histone code”, which regulates the topology of the chromatin, the accessibility of promoters and, therefore, regulates the transcriptional activity and other processes such as replication and DNA repair.4,79 Some histone modifications associated to open chromatin and, therefore, to transcriptional activation, are lysine (K) acetylation in histone H3 (H3K4, H3K14, H3K9, H3K27) and histone H4 (H4K5, H4K20) (Figure 1).
The enzymes in charge of histone acetylation are the HATs. Methylation of lysine catalyzed by HMTs can activate or repress transcription; for example, monomethylation of H3K4, H3K79, H3K35 activates transcription. Some marks of closed chromatin and transcriptional repression include the trimethylation of lysine in H3K9 and H3K27 (Figure 1).9 Together with DNA methylation, histone modifications have been implicated in the etiology and progression of diverse types of cancer.80 Mutations in different histone modifying enzymes that lead to the loss or gain of its function have been found. In particular, acetylation removes positive charges in histones, which decreases the interaction of phosphate groups of the DNA (negative charge) with the N-terminal portion of histones (positive charges). The latter leads to relaxation of the chromatin, which is associated with greater DNA accessibility and, therefore, increased transcriptional activity.81 HDACs enzymes include the HDAC1, HDAC11 and sirtuins.82 Reduction in histone acetylation derived from the overexpression of HDACs enzymes is a common event in diverse tumors, including leukemia.81–84
In pediatric ALL, overexpression of histone deacetylases, such as HDAC1, HDAC2, HDAC3, HDAC6, HDAC7 y HDAC8, has been identified in comparison to non-leukemic cells (Figure 2).84,85 Particularly, overexpression of HDAC2 and HDAC5 in ALL-B has been reported,84,86 whereas in ALL-T a greater expression of HDAC1, HDAC4 and HDAC5 has been observed.84 As for prognosis, overexpression of HDAC1, HDAC2, HDAC3, HDAC4, HDAC7, HDAC9 y HDAC11 genes has been associated with an unfavorable prognosis in pediatric ALL.84,85,87 Furthermore, acetylation of the H4 histone has been proposed as a marker of prognosis at diagnosis and in relapses. In adults with ALL, increased levels of H4 acetylation correlated with an increase in survival.88 Additionally, ETV6-RUNX1 has been reported to recruit HDACs in ALL-B, which induces remodeling of the chromatin and, consequently, blocks the transcription of certain genes normally activated by RUNX1.89
Although not all the signaling pathways and cell processes in which the alteration of HDACs have been identified in pediatric ALL, they are known to be involved in processes such as transcription (HDAC1, HDAC3, HDAC4, HDAC6, HDAC7, HDAC8, HDAC9), cell cycle regulation (HDAC1, HDAC2, HDAC3, HDAC4, HDAC6, HDAC7), apoptosis (HDAC1, HDAC2, HDAC3, HDAC6, HDAC7), and p53 (HDAC2) and Notch (HDAC1, HDAC3, HDAC7) signaling pathways (Figure 3).4
In ALL, altered HATs also have been identified. The main HATs include the GNAT MYST, and CBP/p300 families.82 Some of these proteins have been involved in chromosomal rearrangements capable of leading to leukemic transformation through alteration of histone acetylation and, consequently, in gene expression.90 Some HATs that have been identified to be overexpressed in ALL-B include KAT7, KAT2A, CREBBP (CPB or KAT3A) and KAT6B.86 This modification has been associated with cell processes such as transcription regulation, proliferation and apoptosis.86 Additionally, it has been observed that KAT2A acetylates and stabilizes the oncoprotein TCF3-PBX1 in ALL cells.91 It has also been found that mutations in the CREBBP gene, which lead to a transcriptional deregulation of its target genes, are associated to pediatric ALL-B relapse.92,93 It has been determined that the overexpression of CREBBP can confer a bad prognosis, including those ALL-B subtypes with standard risk, such as hyperdiploidy.93
The most studied HMT in ALL is MLL, which methylates lysine 4 of the H3 histone (H3K4), which is a mark of open chromatin and, therefore, favors transcription.4 Additionally, MLL regulates transcription through recruitment of HAT protein such as CBP and MOF. HOXA genes are mainly part of MLL multiple targets. The MLL forms rearrangements with over 85 different genes, which lead to the formation of chimeric proteins. Although they lose the active methyltransferase H3K4 domain, they retain the capacity of binding to chromatin.94,95 MLL oncoproteins lead to the aberrant activation of their target genes through diverse mechanisms. For example, the chimeric protein MLL-AF4 alters the regulation of its target genes by recruiting epigenetic regulation complexes such as SEC and DOT1 (H3K79 methyltransferase). This dysregulation leads to leukemic transformation since MEIS1, RUNX1, FLT3, MYC, BCL2, y PROM1 genes are among the targets of MLL-AF4.96 Moreover, MLL has been found fused to HAT proteins, such as CREBBP and EP300, leading to the overexpression of their target genes.96,97
5Alteration in miRNAsMicroRNAs (miRNAs) constitute another type of epigenetic regulation. They are small RNAs of 18-25 nucleotides which mainly inhibit the translation of their target mRNAs through interaction with the 3′UTR in mammals. miRNAs derive from the processing of precursors by the RNAse III-Drosha-Dicer protein complex.98 Gene expression regulation by miRNAs is a common biological process; more than 60% of mRNAs can be regulated by miRNAs.99 miRNAs are implicated in critical biological processes, including growth and cell development, metabolism, proliferation, differentiation, and apoptosis.17 In cancer, it has been observed that miRNAs participate acting as oncogenes (oncomiRNAs), tumor suppressors, and metastasis suppressors or activators.17 Additionally, miRNAs can be regulated by methylation of the DNA and histone modification (Figure 2).100
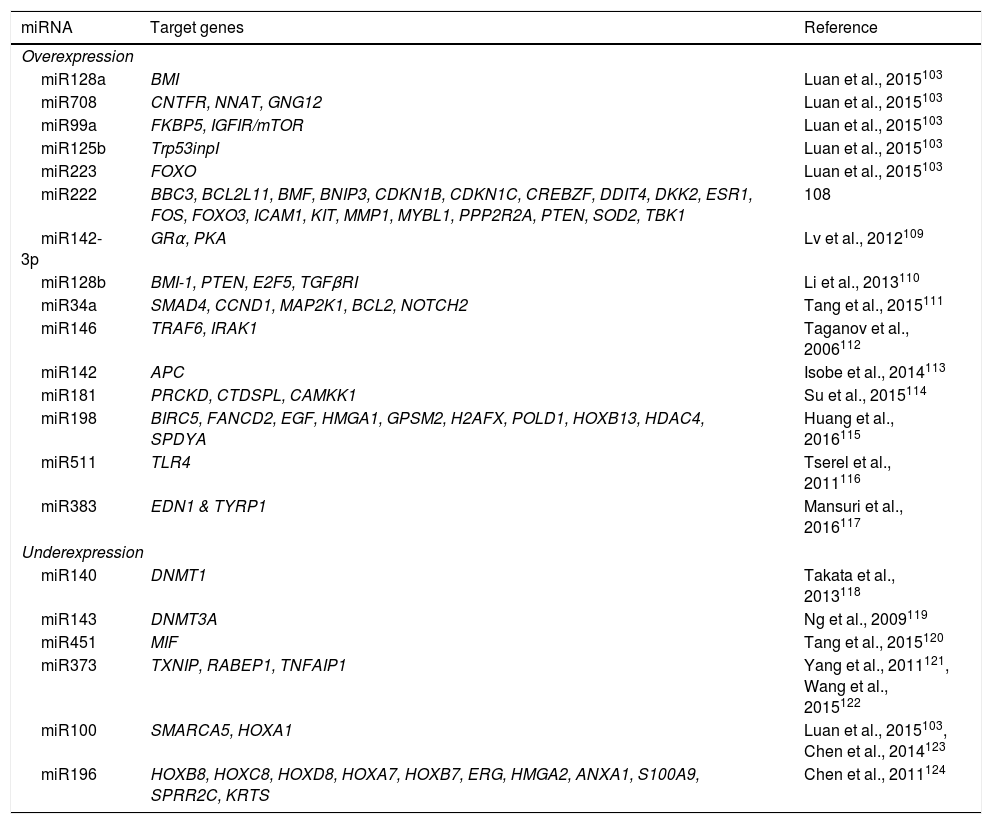
Multiple biological processes are regulated by miRNAs, including normal and malignant hematopoiesis.101,102 Alterations in miRNA expression have been identified in ALL-B and T in both children and adults.17 miRNA expression profiles have been used in the diagnosis, classification, and prognosis of ALL.103 In pediatric ALL, overexpression of miR222, miR339, miR142-3p, miR128a, miR128b, miR34a, miR146, miR142, miR181, as well as decreased expression of miR140, miR143, miR451, miR373, miR100, miR196b has been reported in comparison to their normal cell counterparts (Table 6). Distinctive miRNA signatures between ALL genetic subtypes have also been reported. In the hyperdiploid subtype, overexpression of miR198, miR222, miR223, miRNA511 and miRNA708 has been detected, whereas the ETV6-RUNX1 subtype presents miR99a, miR100, miR125b, miR383 and miR708 overexpression (Table 6).103–124 ALL subtype with MLL rearrangements is characterized by the overexpression of miR196B and low expression of miR128b and miR221.103,104
Target genes of frequently altered miRNAs in ALL.
| miRNA | Target genes | Reference |
|---|---|---|
| Overexpression | ||
| miR128a | BMI | Luan et al., 2015103 |
| miR708 | CNTFR, NNAT, GNG12 | Luan et al., 2015103 |
| miR99a | FKBP5, IGFIR/mTOR | Luan et al., 2015103 |
| miR125b | Trp53inpI | Luan et al., 2015103 |
| miR223 | FOXO | Luan et al., 2015103 |
| miR222 | BBC3, BCL2L11, BMF, BNIP3, CDKN1B, CDKN1C, CREBZF, DDIT4, DKK2, ESR1, FOS, FOXO3, ICAM1, KIT, MMP1, MYBL1, PPP2R2A, PTEN, SOD2, TBK1 | 108 |
| miR142-3p | GRα, PKA | Lv et al., 2012109 |
| miR128b | BMI-1, PTEN, E2F5, TGFβRI | Li et al., 2013110 |
| miR34a | SMAD4, CCND1, MAP2K1, BCL2, NOTCH2 | Tang et al., 2015111 |
| miR146 | TRAF6, IRAK1 | Taganov et al., 2006112 |
| miR142 | APC | Isobe et al., 2014113 |
| miR181 | PRCKD, CTDSPL, CAMKK1 | Su et al., 2015114 |
| miR198 | BIRC5, FANCD2, EGF, HMGA1, GPSM2, H2AFX, POLD1, HOXB13, HDAC4, SPDYA | Huang et al., 2016115 |
| miR511 | TLR4 | Tserel et al., 2011116 |
| miR383 | EDN1 & TYRP1 | Mansuri et al., 2016117 |
| Underexpression | ||
| miR140 | DNMT1 | Takata et al., 2013118 |
| miR143 | DNMT3A | Ng et al., 2009119 |
| miR451 | MIF | Tang et al., 2015120 |
| miR373 | TXNIP, RABEP1, TNFAIP1 | Yang et al., 2011121, Wang et al., 2015122 |
| miR100 | SMARCA5, HOXA1 | Luan et al., 2015103, Chen et al., 2014123 |
| miR196 | HOXB8, HOXC8, HOXD8, HOXA7, HOXB7, ERG, HMGA2, ANXA1, S100A9, SPRR2C, KRTS | Chen et al., 2011124 |
Diverse miRNAs implicated in cell proliferation and apoptosis have been associated with prognosis in patients with ALL. Low expression of miR456, miR708, miR210, as well as overexpression of miR100/99 have been associated with chemotherapy resistance and, therefore, with an unfavorable prognosis. Other miRNAs which low expression is associated with a bad prognosis include miR124a and miR152. Additionally, overexpression of miR92a is associated with an unfavorable prognosis.60,65,125–127
6Epigenetic therapy in ALLThe identification of epigenetic alterations in ALL has motivated the search of molecules capable of reverting these alterations. In patients with ALL, two hypomethylating agents with previous approval in the United States for their use in other types of cancer have been tested. In patients with refractory ALL or in relapses, inhibitors of the DNA-methyltransferase, such as 5-azacitidine and decitabine, in combination with other conventional chemotherapeutic drugs have been used. In these studies, evidence of clinical activity without excessive toxicity was observed.128,129 In addition to demethylating agents, histone inhibitory agents, such as vorinostat and panobinostat, have been tested in patients with ALL. In a phase II study, the administration of decitabine in combination with vorinostat was tested before reinduction chemotherapy in children and adults with refractory ALL. In one out of 13 patients, death by toxicity was registered. In 5/8 patients who completed the treatment, an allogeneic hematopoietic stem cell transplantation was performed, of whom three died of transplant related causes and two survived without evidence of the disease.130
Additionally, it has been proposed that through the regulation of certain miRNAs, an improvement of the therapeutic effect of glucocorticoids could be achieved, particularly for those patients that show resistance to agents such as prednisone. It has been demonstrated that miRNA-17 inhibition leads to an increase in the sensitivity of leukemic cells to dexamethasone.131 In those patients with MLL rearrangements insensitive to glucocorticoids treatment, restitution of the expression of miR128b and miR221 has been suggested as therapy. Both miRNAs show low expression in ALL patients with MLL rearrangements. It has been observed that miR128b has MLL, AF4 genes as targets, and even the oncogenic fusions MLL-AF4 and AF4-MLL. Moreover, miR222 is capable of negatively regulating CDKN1B. In a cooperative manner, both miRNAs are capable of potentiating the sensitivity of leukemic cells to glucocorticoids.132 The restoration of the expression of miR143 has also been proposed as therapy for patients with ALL with the MLL rearrangement; in these patients, miR143 is hypermethylated and silenced. This miRNA has been identified as a regulator of the expression of MLL-AF4. miRNA143 restitution induces apoptosis of leukemic cells.133
In patients with BCR-ABL1-positive ALL, restitution of miR203 has been proposed as therapy. This miRNA has ABL1 and BCR-ABL1 as targets. In BCR-ABL1-positive leukemia, miR203 has been found to be silenced by genetic and epigenetic mechanisms. In patients with BCR-ABL1-positive ALL, who show resistance to tyrosine-kinase inhibitor, it has also been suggested that restitution of miR217 could be used as therapy to inhibit DNTM3A.134,135 It has been reported that patients with BCR-ABL1-positive ALL acquire resistance to tyrosine-kinase inhibitor through the downregulation of miR217, which in turn up-regulates DNMT3A.
7ConclusionsEpigenetic regulation through DNA methylation, histone modification, and by miRNAs plays a fundamental role in the development and evolution of ALL and other types of cancer. In ALL, these levels of regulation have been involved in the etiology of the disease, as well as in the diagnosis, classification, and prognosis of patients. The advent of genome wide association studies allows the detection of an ample number of epigenetic modifications characteristic of ALL, which reflect the complexity and heterogeneity of epigenetic regulation in this disease. Research in this field will keep offering information to allow a better understanding of the complex etiology of ALL. Moreover, knowledge of the epigenetics of ALL will contribute to identify therapeutic targets that could be modified with specific treatments, with the advantage that, unlike genetic modifications, epigenetic alterations are reversible.
Conflict of interestsThe authors declare no conflict of interests of any nature.
Miguel Alemán 2012 Foundation, Federal Funds 2013 Instituto Nacional de Pediatría, Sectorial Fund of Research for Education SEP-CONACyT CB-2012-01/183467. Navarrete-Meneses thanks to the Biological Sciences Postgraduate Program, CONACyT 385279.
Please cite this article as: Navarrete-Meneses MdP, Pérez-Vera P. Alteraciones epigenéticas en leucemia linfoblástica aguda. Bol Med Hosp Infant Mex. 2017;74:243–264.