














1. Summary of the clinical history (A-13-05)
We present the case of a female patient referred in 2008 at 5 months of age to the Hospital Infantil de México Federico Gómez (HIMFG) with a diagnosis of bilateral congenital cataract.
1.1. Family history
The patient’s mother is a 29-year-old housewife and the 35-year-old father is a farm worker. The patient has a half-brother 12 years of age. Consanguinity between parents was probable.
1.2. Non-medical history
There is no significant history; all are residents of the State of Mexico.
1.3. Perinatal and medical history
The mother had a history of spontaneous abortion of unknown etiology. The patient was the product of a third pregnancy and the mother received iron, folic acid and tetanus toxoid vaccine. Four ultrasounds were performed, the last of which reported microcephaly. The patient was born at term by cesarean section with a weight of 3180 g, Apgar score 9/9. The mother was discharged from the hospital.
The patient had microcephaly, central hypotonia and psychomotor delay. Auditory potentials indicated bilateral moderate hypoacusia. Imaging studies showed hyperdensity of the white substance from the centrum semiovale and bilateral periventricular with isolated calcifications and a retrocerebellar arachnoid cyst.
January 2009. Papuloplasty and vitrectomy were performed. That same year, funduplication and gastrostomy were done due to alteration in the swallowing mechanism. Indications for feeding with thickened liquids of 900 kcal were given.
September 2010. A genetic profile and TORCH were requested and the karyotype was 45XX. Diagnosis of Cockayne syndrome was made.
May 2011. Hospitalized for pneumonia. During this hospitalization, the patient received two transfusions of red blood cell concentrate for pneumonia.
May 2013. The patient presented to the emergency department for edema of the hands and feet, irritability, abdominal distention and lack of evacuation. Community-acquired pneumonia was diagnosed. Hemoglobin of 4.8 mg/dl and hematocrit of 16% was found with shock for which red blood concentrate was transfused. Management with amines for 24 h was initiated and antibiotic coverage with cefepime and amikacin. The patient was discharged in an improved condition.
1.4. Current illness
The patient presented to the emergency department for respiratory difficulty and neurological deterioration. On admission she was found to be hemodynamically unstable, with low output, 4-sec capillary refill, weak pulses and tachycardia. Four saline boluses were given at 20 ml/kg and the patient was intubated with a #4.5 cannula. She persisted with systemic inflammatory response and metabolic acidosis for which two more saline boluses were given and adrenaline infusion at 0.3 μg/kg/min.
ArteriAl blood gAs. pH 7.22, pO2 64 mmHg , pCO2 40.5 mmHg, oxygen saturation 87.6%, HCO3 16.2 mEq/l, lactate 1.8 mmol/l, arteriovenous oxygen difference 5.58, Kirby index 64.
Blood count. Leukocytes 15,700/µl, neutrophils 67%, bands 10%, lymphocytes 1%, hemoglobin 8.2 g/dl, hematocrit 25%, platelets 388,000/µl.
Blood chemistry. Urea nitrogen 34 mg/dl, creatinine 1.3 mg/dl, glucose 89 mg/dl, LDH 524 U/l, phosphorus 5.2 mg/ dl, calcium 6.3 mg/dl, sodium 139 mEq/l, potassium 4.6 mEq/l, chloride 105 mEq/l, total bilirubin 0.92 mg/dl, direct bilirubin 0.55 mg/dl.
After the appearance of tachyarrhythmia, adrenaline was changed to dobutamine and infusion of amiodarone was begun at 5 μg/kg/min. The patient continued with persistent hypoxia, venous reserve of 17%, poor perfusion and desatu-ration of up to 39% despite ventilatory support. She finally presented bradycardia and asystole. Advanced resuscitation maneuvers were initiated with ten cycles of compressions, four doses of adrenaline and one dose of bicarbonate. There was fresh blood coming from the endotracheal cannula and she was declared dead.
2. Case presentation
2.1. Imaging (Dra. Pilar Diez)
X-ray of pelvis from 2009, at 1 year of age, showed left hip dysplasia (Figure 1A). Acetabular angles measured 28-29° at birth (Figure 1B). At 1 year of age this angle should have decreased to at least 22°, as seen in the right hip, although this was not shown on the left hip that measured 29°. In addition, the femoral head should be in the lower or inner quadrant, which did not occur in the case of the left hip. The hip morphology is altered. It is flat, the obturator openings are not seen and there is agenesis of the coccyx (Figure 1C).
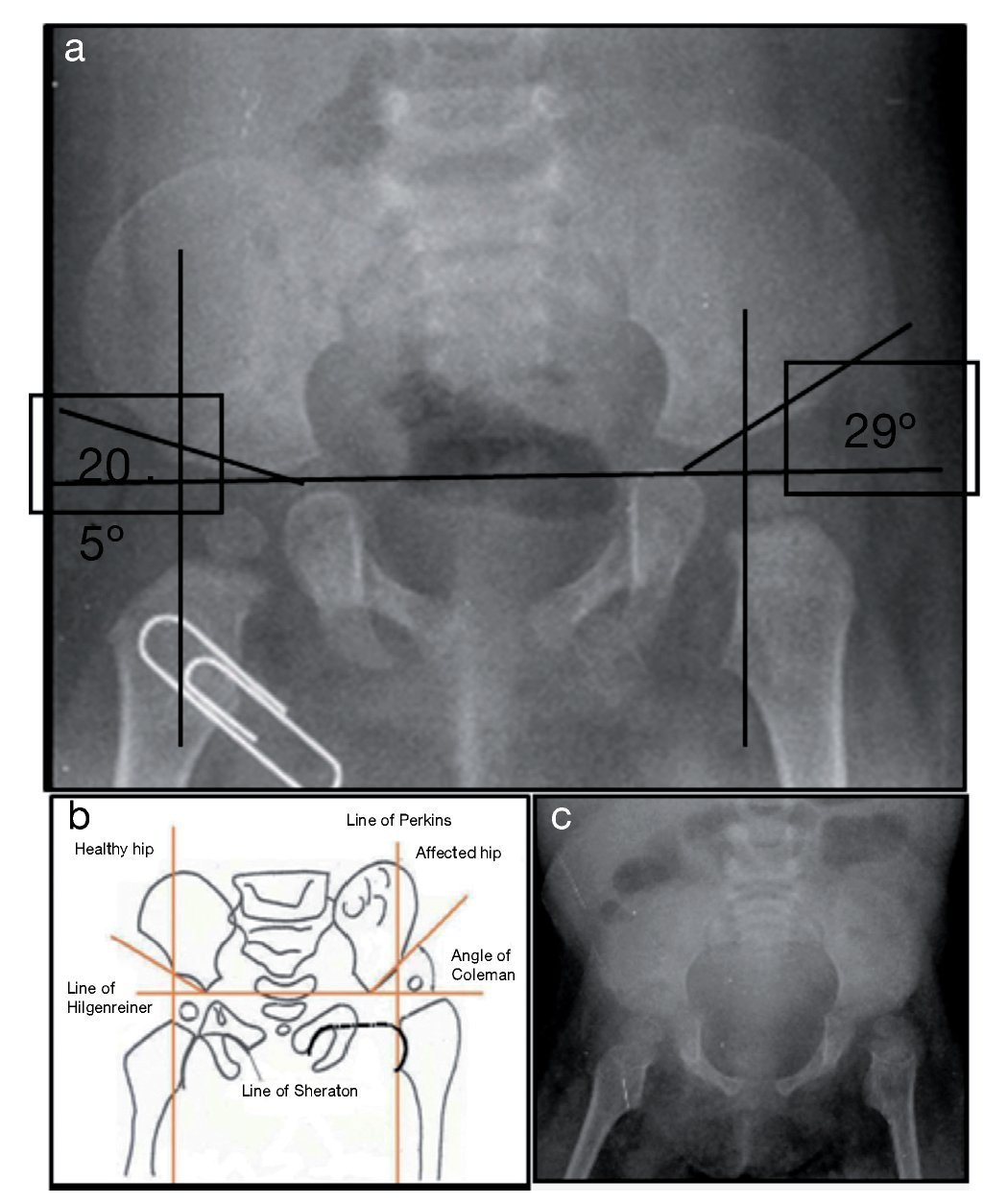
Figure 1. Left hip dysplasia.
Lateral x-rays of the spine show levoconvex thoracic scoliosis with changes in spine morphology (Figure 2). The thoracic curvature is more pronounced than normal for the patient’s age. Thoracolumbar scoliosis is explained by the patient’s lack of ambulation. Vertebral bodies are discretely flattened, elongated, and with notches. The intersomatic spaces are very increased. There are also changes of the sacrum and the coccyx.
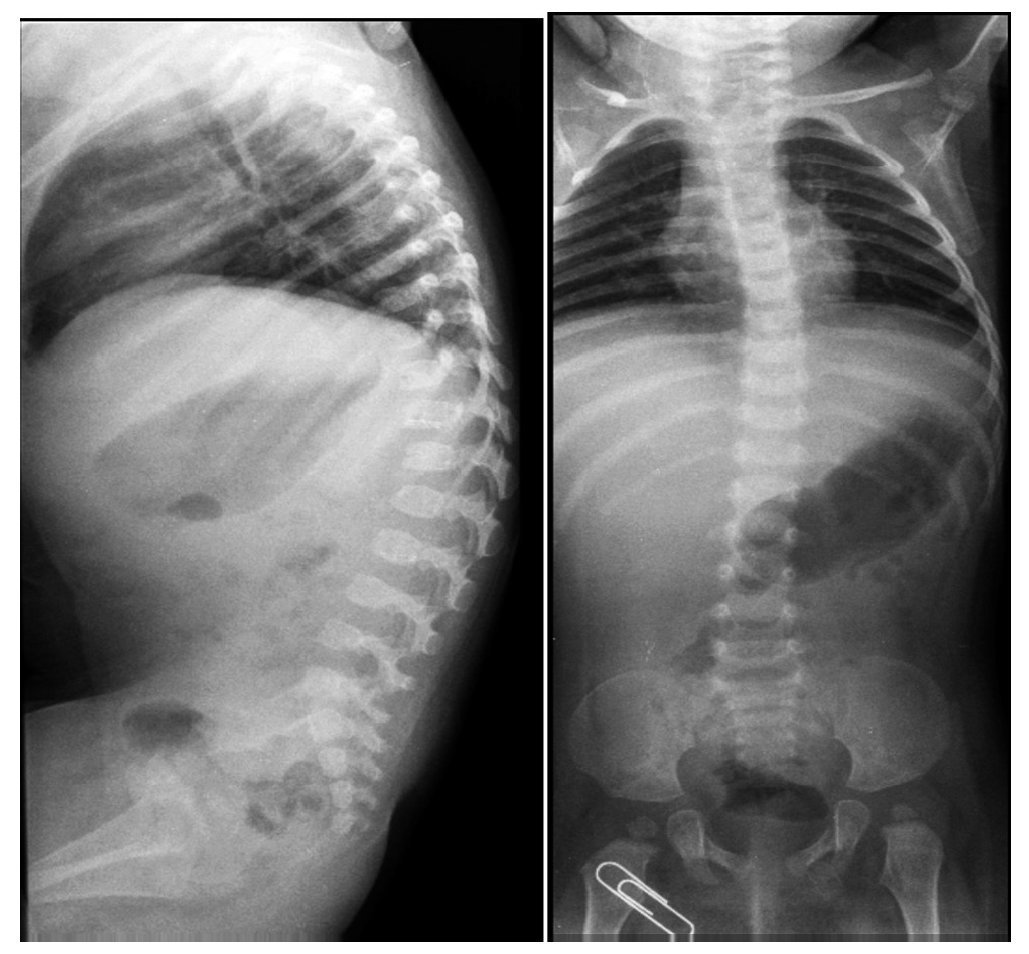
Figure 2. Levoconvex thoracic scoliosis, thoracolumbar xyphosis and alterations of the verterbral morphology.
Esophagogastroduodenal series (Figure 3) showed velo-palatine dysfunction with nasopharyngeal reflux (Figure 3A). The following images show postsurgical changes from a discretely tightened funduplication, which caused a decrease in the caliber of the distal third of the esophagus (Figure 3B), and the latter shows a gastrostomy tube (Figure 3C).
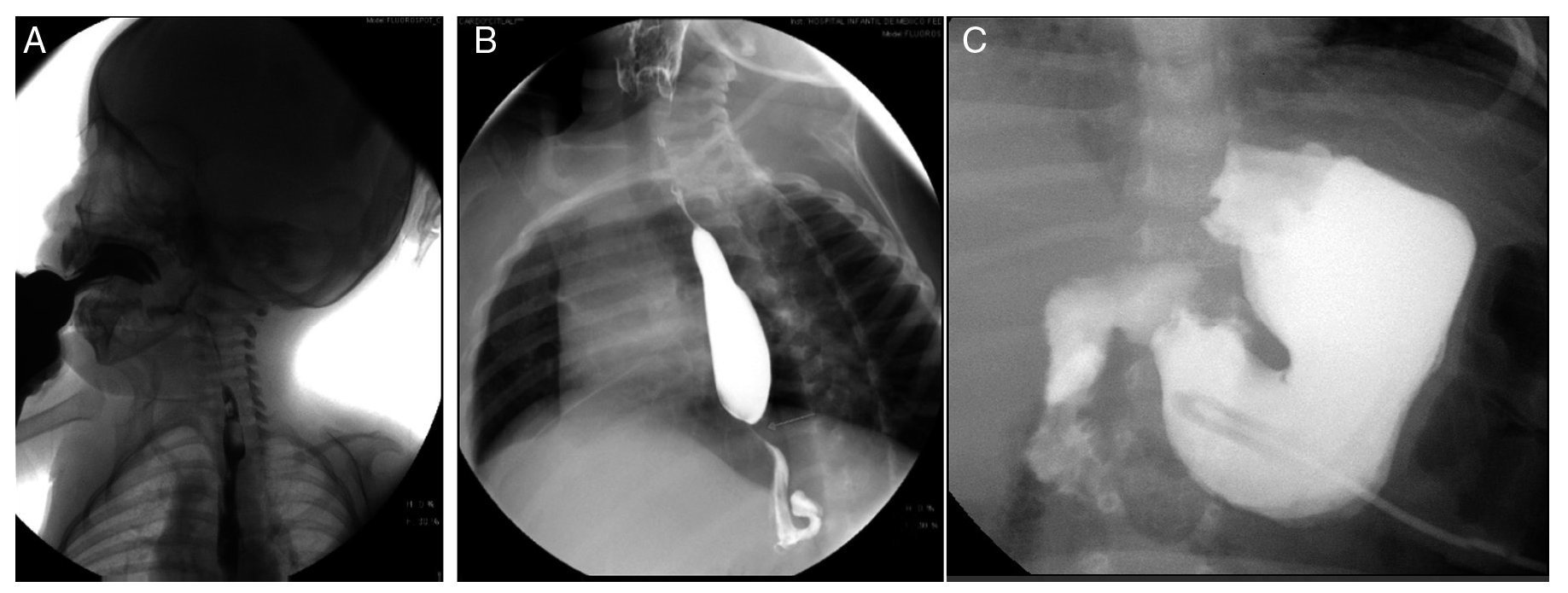
Figure 3. Gastroduodenal esophageal series. (A) Nasopharyngeal reflux (2009). (B) Changes due to funduplication (2011). (C) Gastrostomy tube (2012).
Thoraco-abdominal x-ray showed a reticular nodular infiltrate of the right and left, probably secondary to atelectasis at the location of the endotracheal cannula in the right bronchus (Figure 4). It also showed hepatomegaly and altered vertebral bodies. The ribs and clavicles were thinned with sclerosis at the margins. These images are characteristic of Cockayne syndrome.
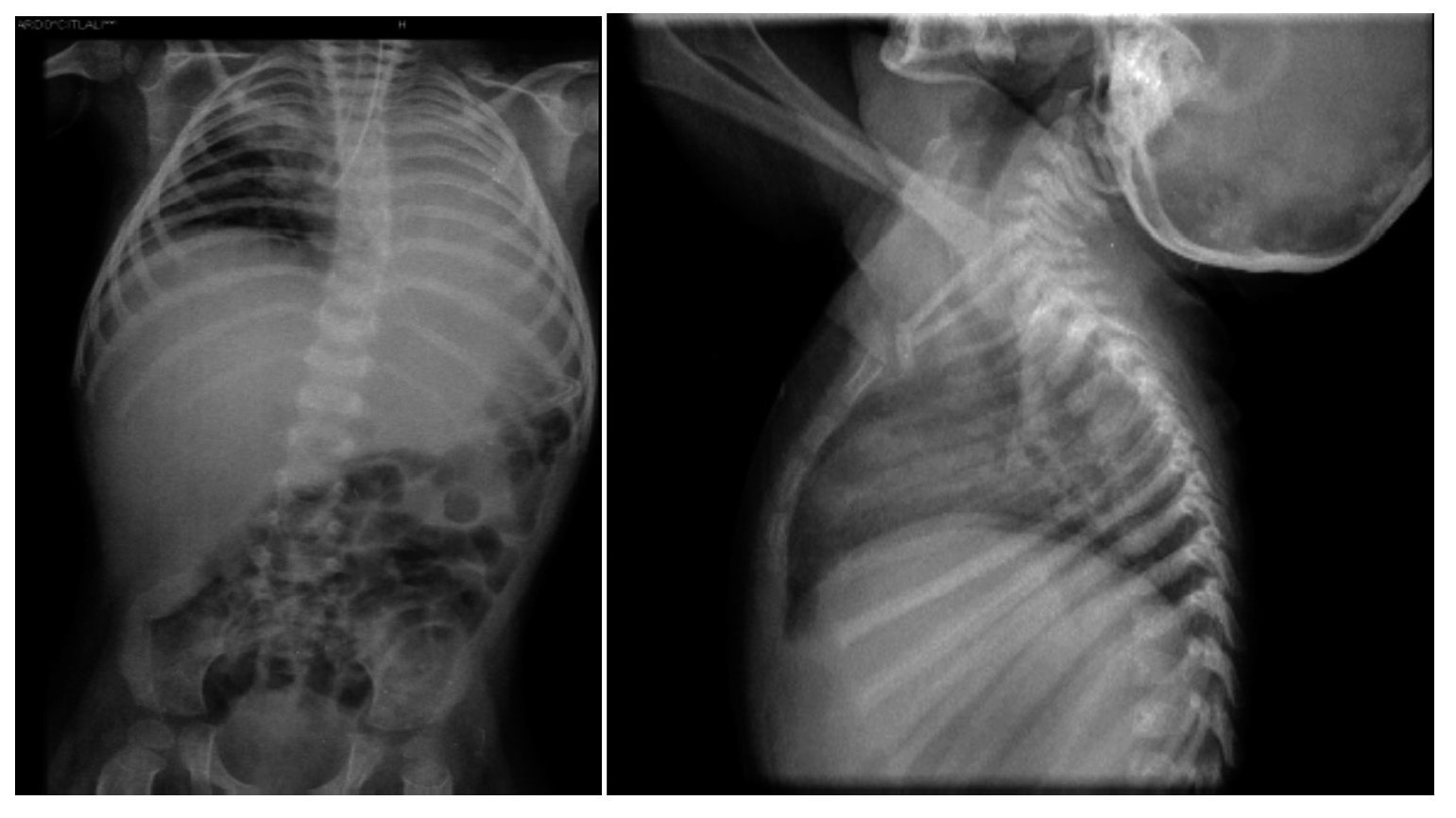
Figure 4. Right reticulo-nodular infiltrate and left atelectasis. Morphological alteration of the ribs, characteristic of Cockayne syndrome.
Facial and skull x-rays showed the nasal bridge in the shape of a hook and hypertelorism. Over the years, the patient’s face loses fatty tissue. The sella turcica is without alterations (Figure 5).
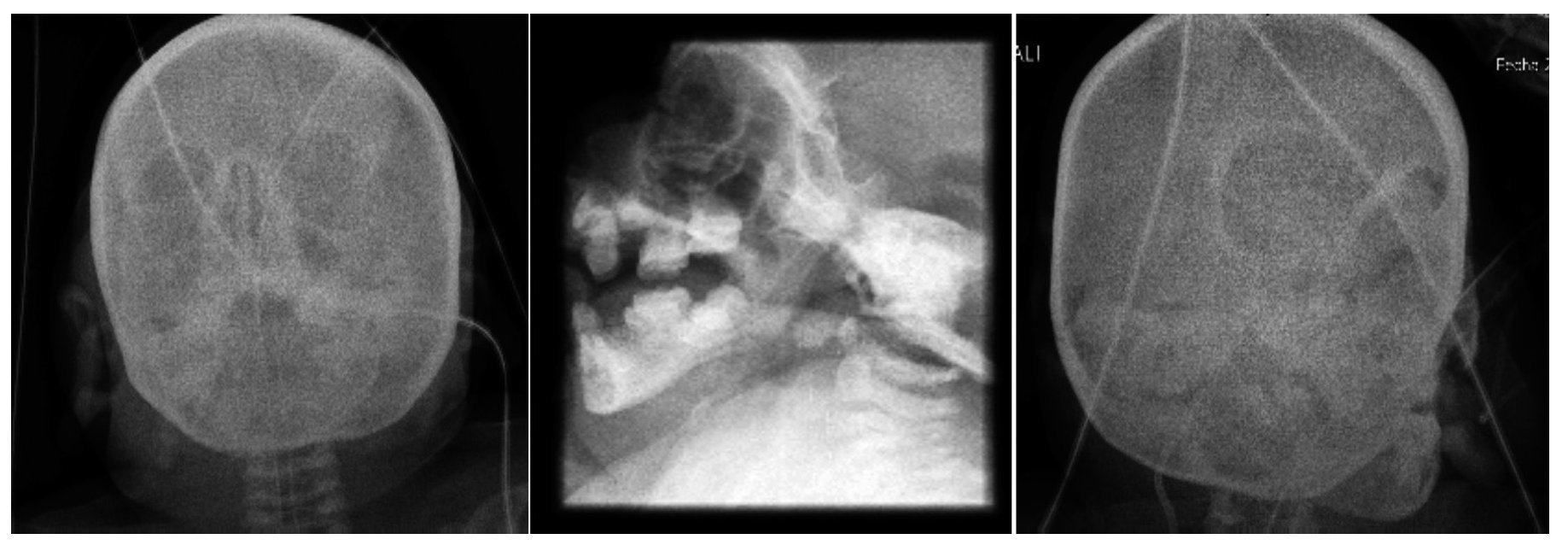
Figure 5. Craniofacial alterations characteristic of Cockayne syndrome.
There were other findings such as short and wide metaphyses and very thin and osteopenic diaphyses with areas of sclerosis (Figure 6).
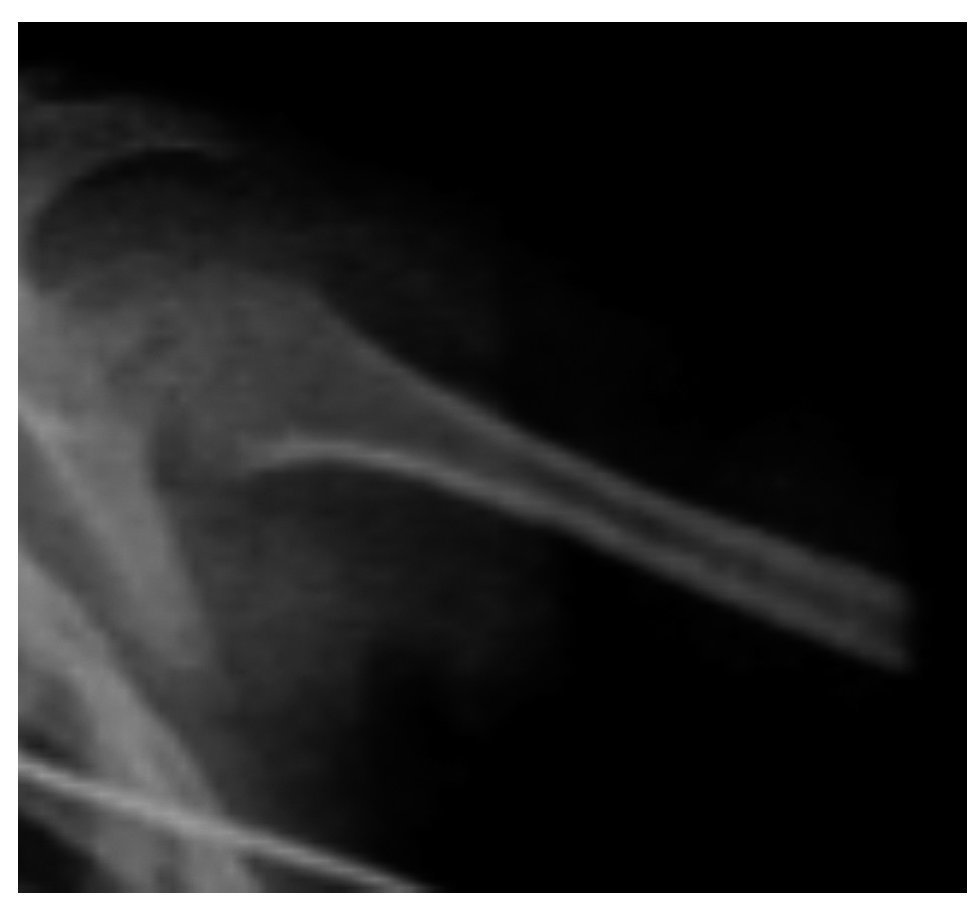
Figure 6. Metaphyses and diaphyses alterations characteristic of Cockayne syndrome.
3. Pathology (Dr. Carlos Alberto Serrano)
Phenotypic findings presented by the body were progeroid facies, thin, dry hair, multiple cerebral calcifications, micro-cephaly (brain weight of 300 g vs. an expected weight of 1200 g) and cerebral atrophy. Sagittal cuts of the brain showed characteristics typical of Cockayne syndrome: almost total loss of the white matter (only gray matter was seen, which gave a thin and elongated appearance to the circumvolutions) with widened grooves and discrete dilatation of the ventricular system. Brown-colored areas that corresponded to microcalcifications were observed. Multiple calcifications in the brain parenchyma and loss of neuronal bodies were observed in the histological sections of the basal nuclei. Residual neurons showed pyknotic nuclei with decrease of the cytoplasm and signs of early cell death as well as Purkinje cells of the cerebellum (Figure 7). In a close-up, poorly defined areas, white and granular in appearance were observed, which corresponded to calcifications in various regions of the cerebral cortex. Histological cuts showed white areas that corresponded to myelin loss, which was corroborated with luxol-fast-blue stain. There are two theories on the loss of white matter in this syndrome: the first corresponds to degeneration of myelin-producing cells and the second, due to vascular changes in the brain, which lead to the death of these cells. Histological features observed in these patients are similar to those observed in elderly patients with thickening of artery walls and even atheroma. These changes were not observed in the patient but there were alterations in the morphology of the endothelial cells conducive to a reduction in irrigation of the cerebral parenchyma (Figure 8).
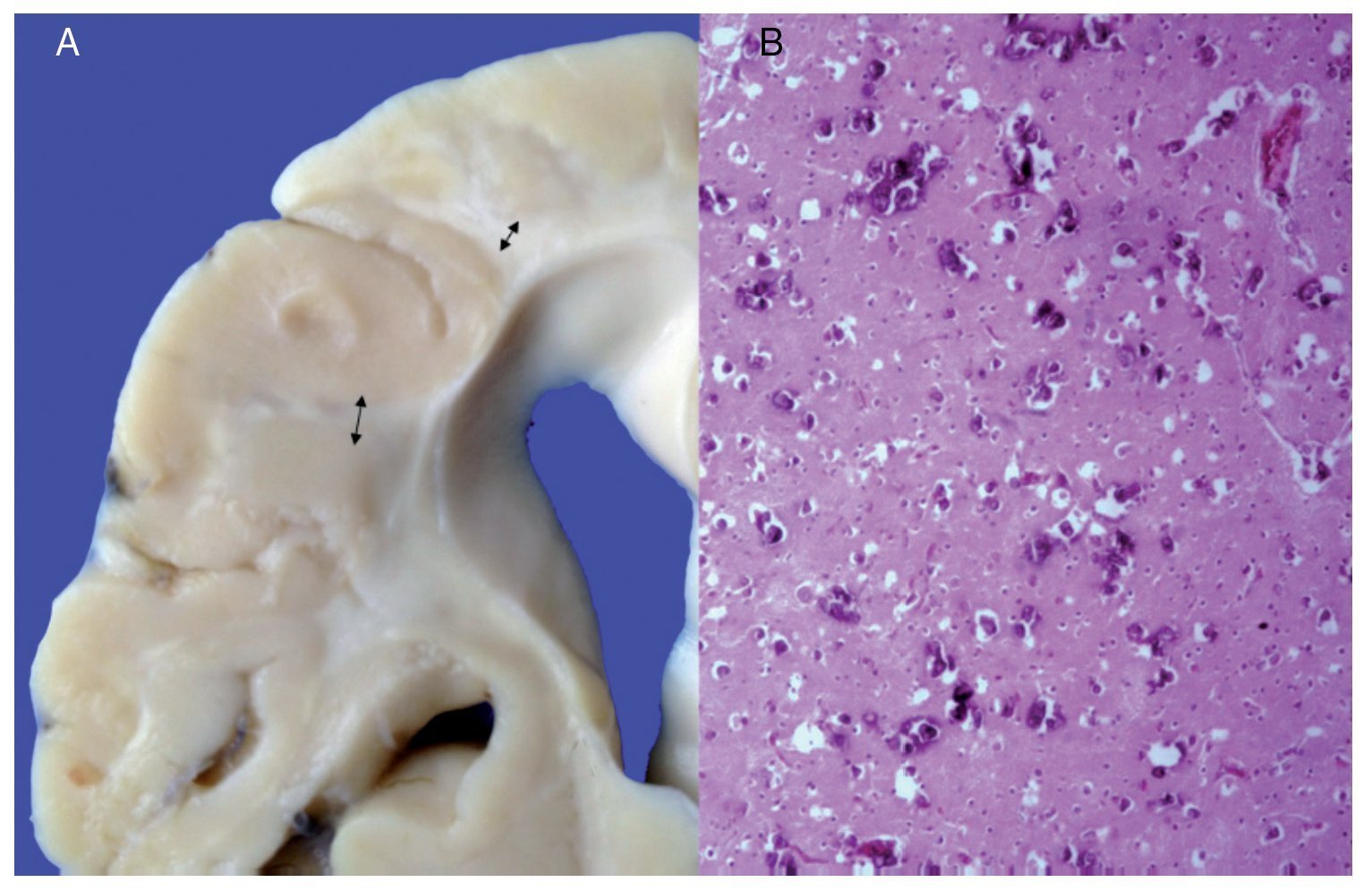
Figure 7. (A) Brain with significant loss of white matter (arrows). (B) Cerebral cortex with multiple calcifications.
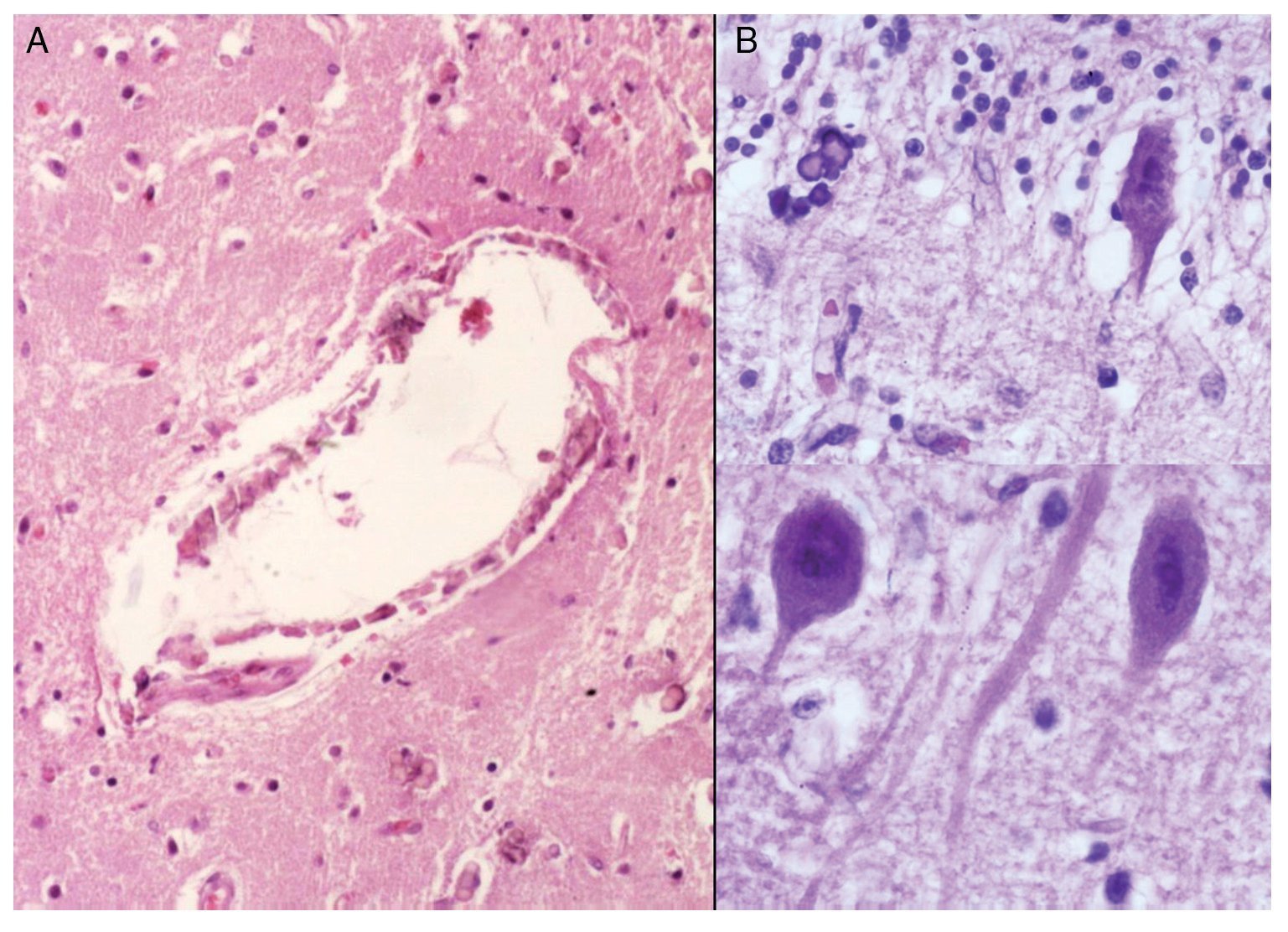
Figure 8. (A) Vessels of cerebral cortex with wall calcifications. (B) Red degeneration of the neurons with microcalcifications.
There was a left pleural effusion of ~75 ml. More than 80% of the pulmonary parenchyma had a dark brown color and the trachea presented areas of mucosal congestion. As can be seen in the histological sections, these dark brown areas correspond to important intra-alveolar bleeding. Eosinophilic fluid was seen in other areas within the alveoli, and acute pulmonary edema was observed. There was also necrosis of the interalveolar septum with formation of hya-line membranes and giant multinucleated cells, indicating chronic bronchial aspiration. Likewise, few lymphocytes and macrophages were observed, which correspond to a resolving pneumonia, but with acute lung damage (Figure 9).
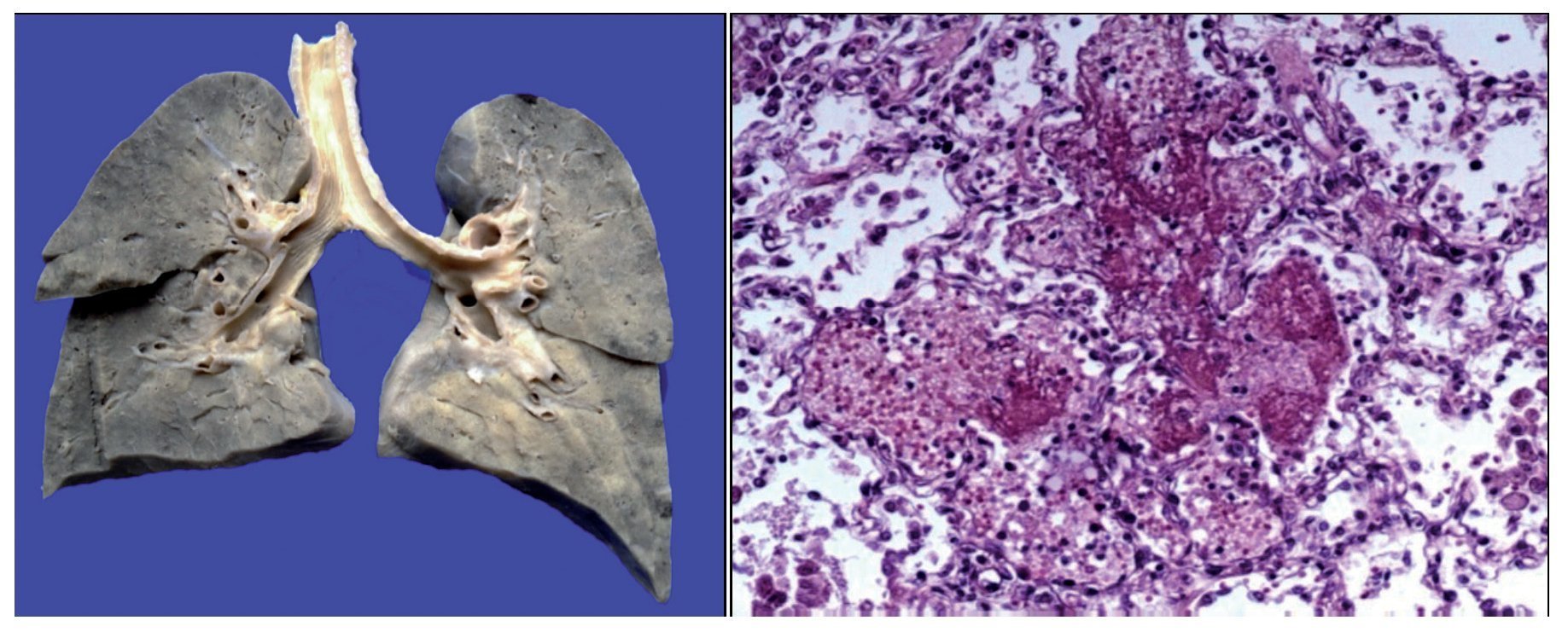
Figure 9. Congestive pulmonary parenchyma with dark brown color. Histological cut with recent hemorrhage and formation of hyaline membranes (acute alveolar damage).
The heart did not show morphological changes but did show changes due to chronic hypoxia characterized by cellular atrophy with shrunken cytoplasm, vacuoles, loss of the nucleus and important intercellular edema.
The esophagogastric junction had thickened folds and mucosal edema. Microscopy showed data of chronic inflammation, hyperplasia of the epithelium and hypoxic changes in the muscular layers. The small intestine and colon only showed hypoxic changes. The liver presented homogeneous, yellowish and micronodular parenchyma in some areas. At the histological level there was macrovesicular steatosis, which is explained by the nutritional status and the state of shock that the patient finally presented (Figure 10).
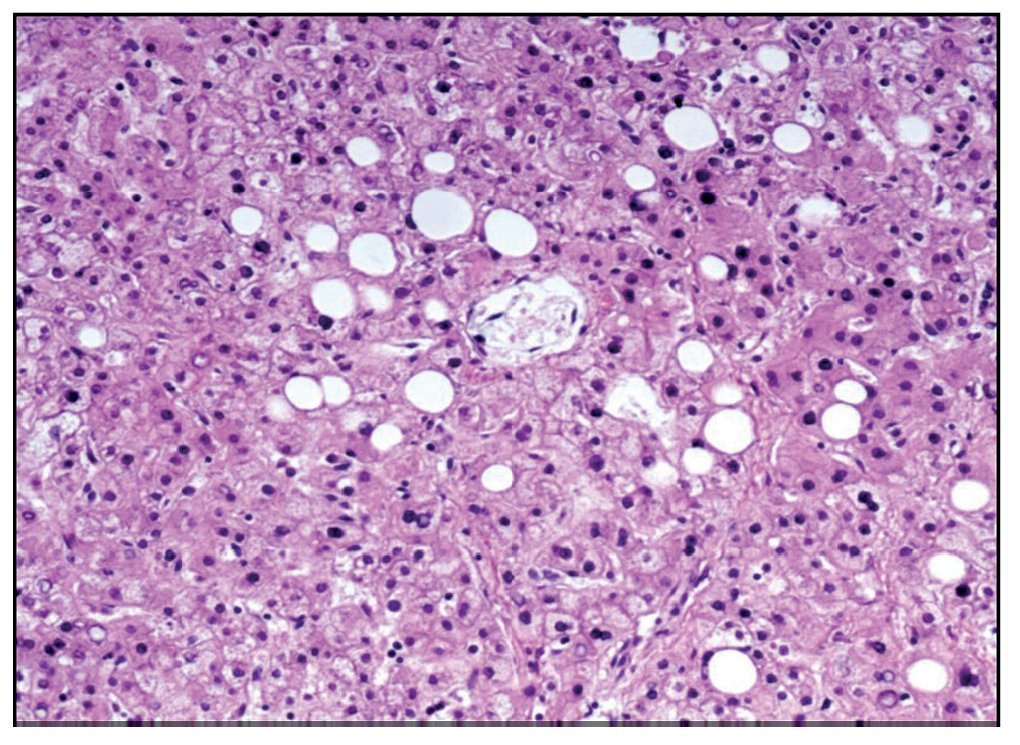
Figure 10. Macrovesicular hepatic steatosis.
The kidneys were discreetly lobulated with congestion in the renal medulla. Cystic dilations of the tubules were demonstrated microscopically with a bit of proteinaceous material in its interior, mainly at the corticomedullary junction. The glomeruli were well preserved (Figure 11).
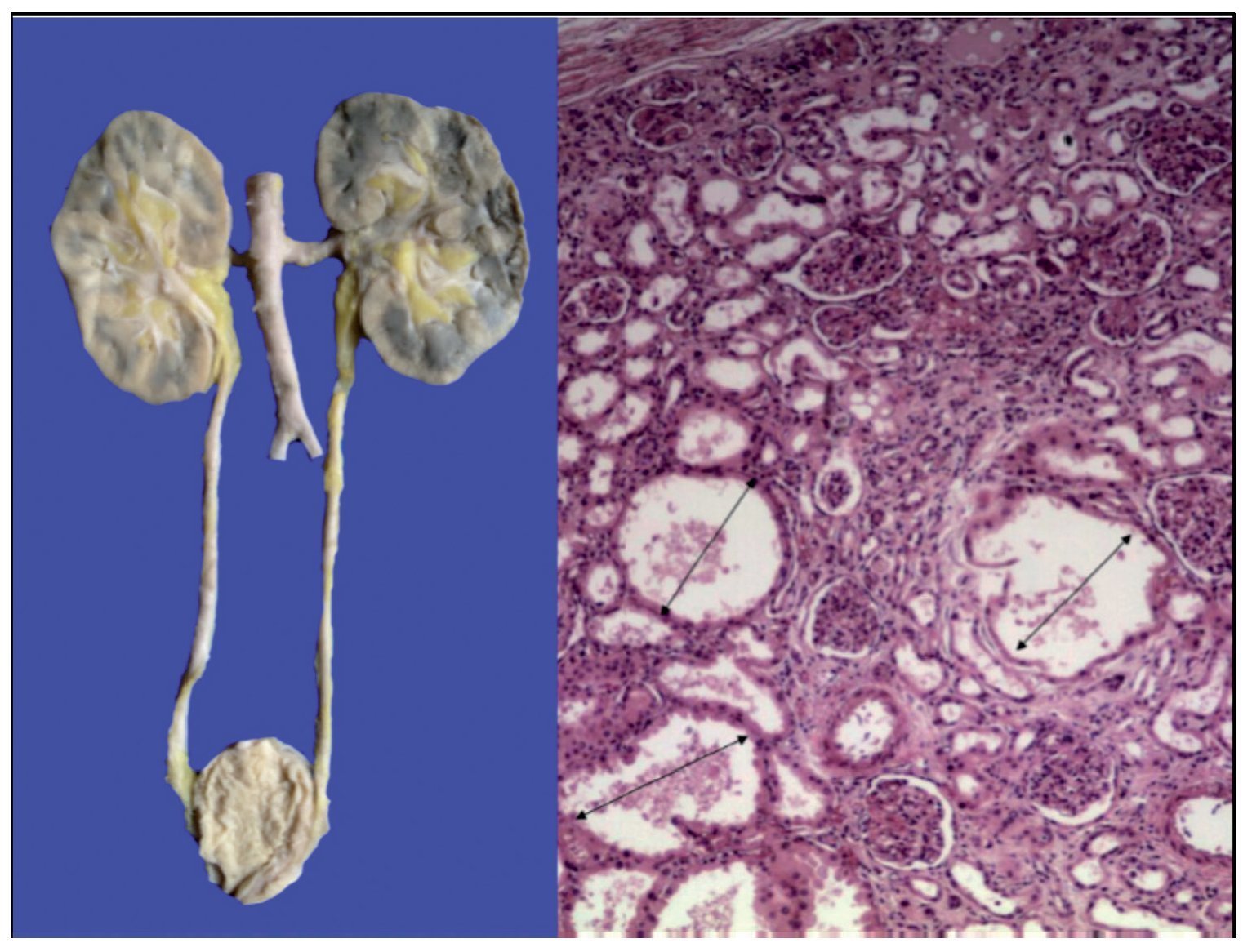
Figure 11. Lobulated kidneys with medullary congestion. Microscopic images show cystic dilatations in the medullary cortical junction (arrows).
Bone marrow had normal cellularity. The three hematopoietic series showed proper maturation. Lymph nodes showed scant lymphoid tissue. In some lymph nodes there is no formation of secondary follicles.
3.1. Final pathological diagnoses
The principal diagnosis in this case was Cockayne syndrome.
3.2. Concomitant disorders
Concomitant pathological diagnoses were as follows:
• Status post funduplication and gastrostomy
• Resolving aspiration pneumonia
• Bilateral pulmonary hemorrhage and extensive diffuse alveolar damage
• Data of anatomic shock: hypoxic-ischemic visceral myopathy throughout almost the entire gastrointestinal system and the myocardium, macro- and microvesicular steatosis in the liver and multivisceral congestion
3.3. Postmortem cultures
Postmortem cultures were negative in all organs with the exception of the colon, which showed Klebsiella pneumonaie. However, there were no histological data of infection.
4. Final comments
Cockayne syndrome is associated with a mutation in the genes (5q11) ERCC6 in 75% of cases and ERCC8 or CKN2 (10q12) in 25% of cases. Both encode for the CS-A and CS-B proteins, which are related to DNA repair.1-3 Changes cause the accumulation of damaged DNA and therefore cellular aging and alterations in cell growth. The cause of death of this patient is explained by demyelination in the central and peripheral nervous system, which caused a swallowing disorder, resulting in long-term chronic bronchial aspiration, multiple episodes of lung inflammation and finally extensive diffuse alveolar damage, leading to death. Changes in DNA repair caused ocular, skin and bone disorders (Figure 12).4-6
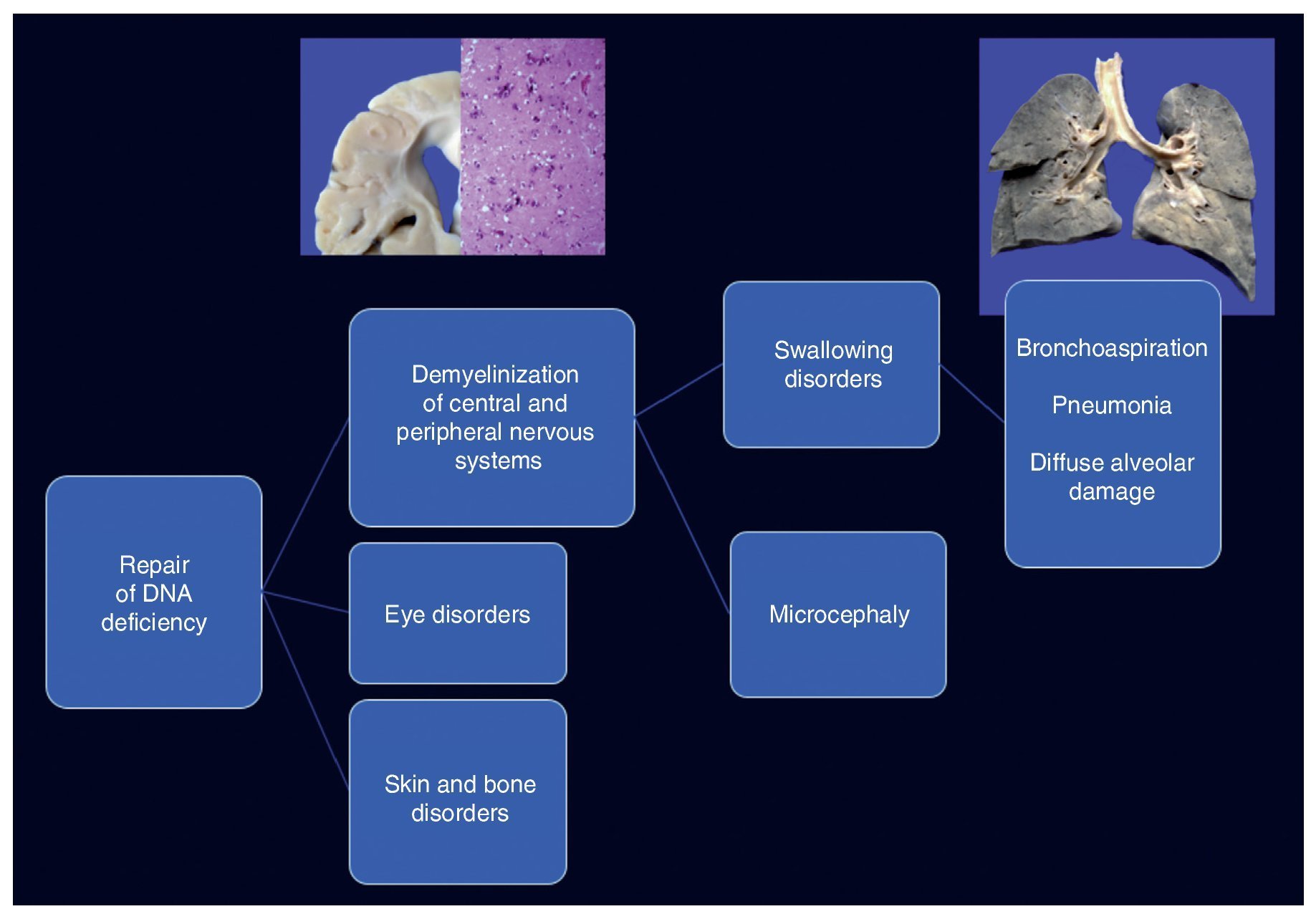
Figure 12. Clinicopathological correlation of the events leading to the death of the patient.
4.1. Genetics (Dra. Alejandra del Pilar Reyes de la Rosa)
There are many reasons for congenital cataracts. Before considering some type of genetic entity, it is necessary to rule out maternal infections during pregnancy. One must also investigate innate errors in metabolism such as galactosemia, which can occasionally present with cataracts from birth. It is important to perform a careful clinical examination to differentiate an isolated congenital cataract from a syndromatic congenital cataract. Within the syndromatic cataracts, there are two large groups: monogenetic and chromosomal disorders.
4.2. Ophthalmology (Dra. Tania Valderrama)
This patient arrived with a total bilateral cataract that would require a multidisciplinary approach involving both pediatrics and genetics. Left eye surgery was done followed by the right eye. An intraocular lens was not placed because patients with systemic disease have more intense inflammatory processes and there is a risk of complications such as glaucoma. The bilateral cataract was total, with metaplasia of the posterior capsule, which is common in patients with systemic syndromes. In addition to the congenital cataract, children with Cockayne syndrome could have enophthalmos such as presented by this patient, retinal dystrophies, strabismus and nystagmus due to poor vision.
4.3. GI nutrition (Dr. Salvador Villalpando)
Due to the finding of severe malnutrition and questions about the opportune time for enteral nutrition alternatives, the importance of taking into account the patient prognosis and for avoiding futile treatment with invasive measures was commented upon.
4.4. Surgery (Dr. Ricardo Ordorica)
In this patient the decision was made to carry out a funduplication and gastrostomy based on the frequency and number of bronchial aspiration episodes she presented and the poor swallowing mechanism. In our study of 96 cases, 75% of patients who had funduplication performed had neurological damage. Whereas this procedure has complications such as esophageal stenosis and accelerated emptying, it improves the nutritional status of the patient along with quality of life.
4.5. Pediatrics (Dra. Saskia Bermúdez)
This is a progressive disease with undesired clinical progression and is associated with multiple co-morbidities. Survival is usually <10 years of age. One has the responsibility to provide parents of these types of patients with genetic counseling as well as information about the fatal prognosis. It is important to discuss the case with an Ethics Committee.
Ethical disclosure
Protection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of data. The authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consent. The authors declare that no patient data appear in this article.
Conflict of interest
The authors declare they have no conflicts of interest.
Received 8 July 2015;
accepted 10 July 2015
* Corresponding author.
E-mail:crls.serrbe@gmail.com (C.A. Serrano).