



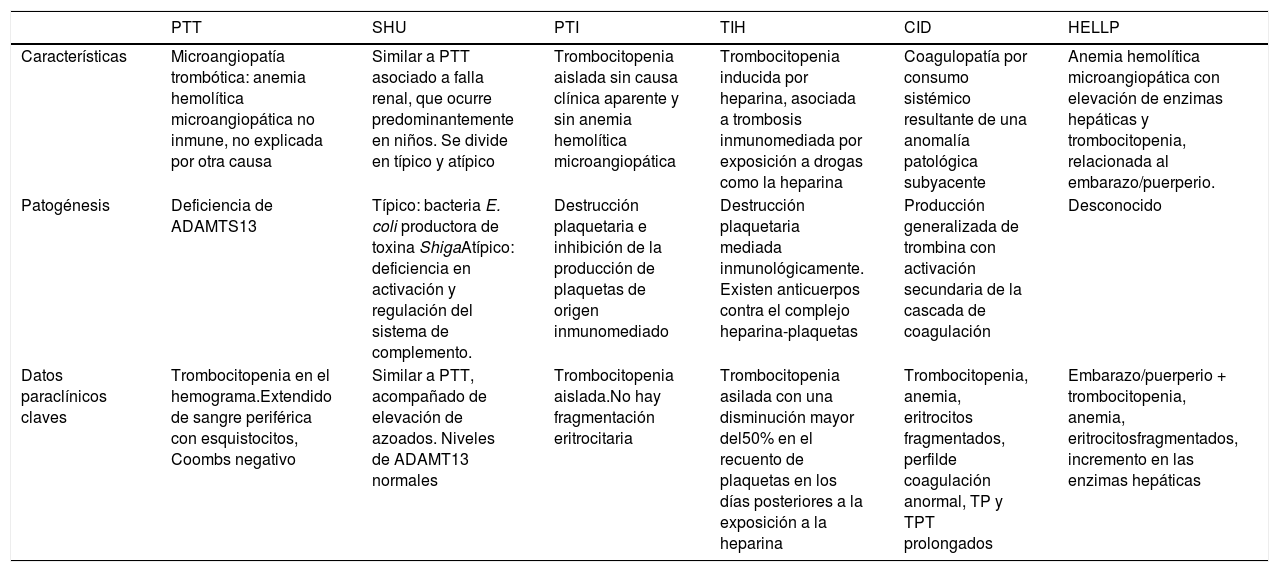
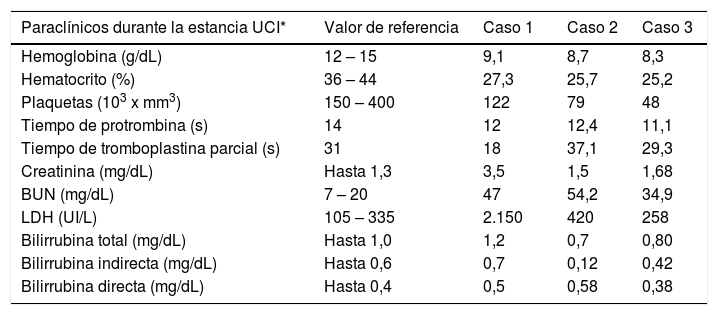
El síndrome hemolítico urémico atípico se caracteriza por anemia hemolítica, trombocitopenia y lesión renal aguda; ocasionado por una alteración en la regulación del sistema del complemento; es parte de las microangiopatías trombóticas (MAT) con un daño vascular a nivel histológico de los capilares. En este documento se reportan tres casos de pacientes de sexo femenino, entre los 25 y 55 años, con distintas manifestaciones clínicas iniciales, pero con alteraciones a nivel hematológico y renal que caracterizan al síndrome hemolítico urémico. Se evidenció trombocitopenia, anemia y alteración renal en todas las pacientes, aunque en grado variable. Se discute sobre las manifestaciones clínicas, las diferencias con la púrpura trombocitopénica trombótica, principal diagnóstico diferencial, haciendo énfasis en la importancia de tener un alto índice de sospecha clínica para el diagnóstico oportuno y el tratamiento.
Atypical haemolytic uraemic syndrome is characterised by haemolytic anaemia, thrombocytopenia, and acute renal injury. It is caused by a disorder in the regulation of the complement system. It is part of the thrombotic microangiopathies with vascular damage at the histological level of the capillaries. Reports are presented on three cases of female patients, with ages ranging between 25 and 55 years, with distinct initial clinical manifestations, but with haematological and renal changes that characterise haemolytic uraemic syndrome. Thrombocytopenia, anaemia, and renal changes were found in all three patients, although to varying degrees. There is also a discussion on the clinical manifestations and the differences in the thrombotic thrombocytopenic purpura (an entity in which a differential diagnosis is made). Finally, the importance of having a high rate of clinical suspicion for timely diagnosis and treatment is also emphasised
Artículo
Socios de la Asociación de Medicina Crítica y Cuidado Intensivo
![]()
Para acceder a la revista
Es necesario que lo haga desde la zona privada de la web de la AMCI, clique aquí

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
