







In this paper, we report a study of the influence of the synthesis method on the properties of mixed metal oxides. For this, the syntheses of the mixed oxide by chemical coprecipitation and modified polymeric precursor (MPP) methods containing Ni2+, Zn2+, Al2+, and Zr4+ were conducted. For this purpose, the samples were characterized by X-ray powder diffraction, which showed defined peaks for the mixed metal oxides (NiO, ZnO, and ZrO2). The interaction of H2 with mixed oxides has been investigated using temperature-programmed reduction (H2-TPR); this result showed that the modified polymer precursor method favored the presence of the peak at elevated temperatures. The N2 physisorption (BET method) showed that the polymer precursor method provided higher specific surface areas and basic site numbers in relation to the coprecipitation method. Thermogravimetric analysis of the systems decomposition revealed their disintegration at ~550 °C and resulted in mixed metal oxides in both methods.
The development of materials suitable for different applications has become of interest in several areas of knowledge since their application depends heavily on their structural properties, which are affected by their synthesis method. Mixed metal oxides can be obtained by the polymeric precursor, coprecipitation [1,2], sol–gel [3], and hydrothermal methods, among others. Polymer precursor and chemical co-precipitation methods are more commonly used because they are cheaper and have essential characteristics for various applications, namely: high surface area [4]; high thermal stability [5]; basic properties (Lewis sites) [6]; dispersed metallic particles, and formation of homogeneous mixed oxide on a nanometric scale [3,8], which are generally referred to in the literature as mixed metal oxides or mixed oxide solutions [8].
The mixed oxides can be applied in several reactions, such as: (i) alkene polymerization; (ii) aldol condensation of aldehydes and ketonas [5]; (iii) dry reforming and hydrocarbons vapor [1]; (iv) methanation; (v) synthesis of methanol and heavier alcohols; (vi) hydrocarbon synthesis (Fischer-Tropsch), and (vii) hydrolysis of n-trilas [9]. The polymeric precursor method, derived from the Pechini method, is considered one of the most popular techniques. This method was originally developed to obtain powders of multicomponent oxides and dielectric materials with a controlled stoichiometry, which is extremely difficult to obtain by conventional methods [10]. The polymer synthesized is derived from the reaction between metal cations (dissolved in aqueous solution) with carboxylic acids (citric acid), polymerized by a polyalcohol (ethylene glycol). This method promotes a better cations distribution in the atomic level of the polymeric structure [11,12].
As far as the coprecipitation method is concerned, there are numerous works in the literature employing this method in two different ways: coprecipitation at constant pH and coprecipitation at variable pH [7,8]. The most commonly used apparatus for obtaining this compound is called an automatic titrator. This system consists of a reactor, a titrator with constant flow, a pH controller and a temperature controller [13,14]. The coprecipitation method was performed for the first time by Feitknecht in obtaining hydrotalcite. The synthesis of the Mg-Cr-CO3 system was performed starting from the diluted solutions (0.01–0.1 mol L−1). During the extended washing step, the initially amorphous precipitate began to crystallize into a low-order precursor [15]. This method was perfected by Reichle in 1930, allowing a considerable variability in the nature of the cations, besides the control of the morphology and the specific surface area. In Reichle method, the aqueous solutions of the cations (Al/Mg) and the precipitants (Na2CO3/NaOH) were added rapidly with stirring at room temperature. The number of moles of sodium hydroxide used was twice that of magnesium and three times that of aluminum. Promptly, the obtained gel was left under heating for a long crystallization period (18 h). In this method, several factors can influence the synthesis, for example, the speed of addition of one solution over the other, the final pH of the suspension, the degree of agitation, and temperature [16,17].
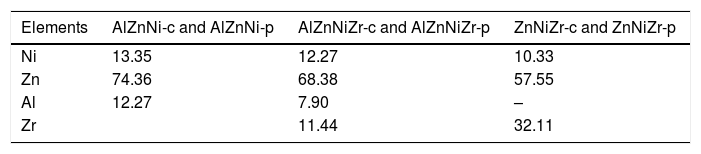
This study aims to conduct a comparative study of the chemical co-precipitation and the modified polymer precursor methods for obtention of mixed oxides. For this, the samples were obtained using cations of Ni, Zn, Al, and Zr, defining the systems under study here, namely AZN-c, AZNZ-c, ZNZ-c, (d) AZN-p, AZNZ-p and ZNZ-p. Subsequently, the precursors were subjected to a thermal treatment at 550 °C to obtain the mixed oxides, defining the systems studied (see Table 1). For structural characterization, X-ray diffraction was employed. Finally, the adsorptive property of mixed oxide was studied with respect to CO2 probe molecule using temperature-programmed desorption.
2Materials and methods2.1Chemicals and materialsRaw materials of zinc nitrate [Zn(NO3)2⋅6H2O], aluminum nitrate [Al(NO3)3⋅9H2O], nickel nitrate [Ni(NO3)2⋅6H2O], zirconium nitrate [Zr(NO3)4⋅5H2O], sodium hydroxide [NaOH], sodium carbonate [Na2CO3], citric acid (C6H8O7⋅H2O) and ethylene glycol (HOCH2CH2OH) were employed as starting materials.
2.2Samples preparation2.2.1Coprecipitation methodThe precursors were prepared by the coprecipitation method [18]. Salts (Ni(NO3)2⋅6H2O, Zn(NO3)2⋅6H2O, Al(NO3)3⋅9H2O and Zr(NO3)2⋅6H2O) were dissolved in 100 ml of deionized water. The formation of the precursor occurred after adding some drops of the metallic salts solution to the precipitating solution (hydroxide carbonate sodium) maintaining the pH ~10.5. The material was placed in a sand bath at 65 °C for 15 h and the resulting slurry was filtered and washed with distilled water until pH ~7 for the removal of residual sodium. The materials were then heated at 100 °C for 15 h and grounded in an agate mortar. Ultimately, the thermal treatment was carried out at 550 °C for 15 h. The calculated mass percentage composition and nomenclatures of the samples are reported in Tables 1 and 2, respectively.
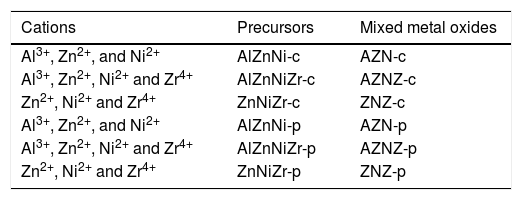
Cations, nomenclature of precursors and mixed metal oxides obtained by coprecipitation and polymeric precursor methods.
| Cations | Precursors | Mixed metal oxides |
|---|---|---|
| Al3+, Zn2+, and Ni2+ | AlZnNi-c | AZN-c |
| Al3+, Zn2+, Ni2+ and Zr4+ | AlZnNiZr-c | AZNZ-c |
| Zn2+, Ni2+ and Zr4+ | ZnNiZr-c | ZNZ-c |
| Al3+, Zn2+, and Ni2+ | AlZnNi-p | AZN-p |
| Al3+, Zn2+, Ni2+ and Zr4+ | AlZnNiZr-p | AZNZ-p |
| Zn2+, Ni2+ and Zr4+ | ZnNiZr-p | ZNZ-p |
The samples were also prepared using a modified polymeric precursor (MPP) route based on the Pechini method [19]. The precursors based on nickel, aluminum, zinc, and zirconium were obtained in accordance with the following steps. First, the separated aqueous solutions of nitrate and citric acid were mixed in a molar ratio of 1:3 metal–citric acid and kept under stirring between 60 and 70 °C. Then, ethylene glycol was added to metal–citric acid aqueous solution in a mass ratio of 1:1 in relation to the citric acid. The gravimetric analysis at 900 °C was performed to determine the amount of each precursor. Thereafter, the polymers were blended according to the desired samples to produce the polymeric precursor. The blends of the polymers were heated to between 80 and 90 °C to form polyester of high viscosity and glass appearance. The polyesters were treated thermally at 300 °C for 2 h to obtain porous powders. These powders were grounded using an agate mortar. Finally, the grounded powders were calcined at 550 °C for 15 h in covered alumina crucibles to obtain mixed metal oxides (see Table 1).
2.3Characterization of the samples2.3.1Thermogravimetric analysisThermogravimetric analyses (TGA) were performed using a Thermobalance Mettler-Toledo under nitrogen flow with a heating rate of 10 °C min−1 until 900 °C.
2.3.2X-ray diffractionX-ray powder diffraction (XRD) was recorded on a Rigaku diffractometer, using Cu-Kα radiation (λ = 1.54056 Å) in the 2θ range between 10° and 80° and with a scanning velocity of 2° min−1.
2.3.3Temperature-programmed reduction of hydrogen (H2-TPR)Temperature-programmed reduction of hydrogen (H2-TPR) was conducted in a Micrometrics Chemisorb 2750 equipment using 10% H2/Ar flowing at 25 mL min−1 with a heating rate of 10 °C min−1. Gaussian-type deconvolution process was performed in all samples [19].
2.3.4Temperature-programmed desorption (TPD) of CO2 (CO2-TPD)The determination of the number and strength of the basic sites was performed by temperature-programmed desorption of CO2 (CO2-TPD) [20]. Firstly, the samples were subjected to a thermal treatment at 550 °C for 0.5 h under He flow (30 cc min−1). After cooling to 35 °C under He flow, the CO2 adsorption was carried out during 0.5 h at 35 °C under CO2 stream. The CO2 was then replaced by He and the TPD started after the baseline stabilization, at the same temperature.
2.3.5Nitrogen physisorptionThe specific surface area was measured by nitrogen adsorption–desorption using a NOVA 1000e QuantachromeInstruments®. Brunauer–Emmett–Teller (BET) equation was employed to calculate the specific surface area. Pore size distributions were calculated from the desorption branch using the BJH method.
3Results and discussionThe temperature-induced degradation profiles for AlZnNi-c, AlZnNiZr-c, ZnNiZr-c, AlZnNi-p, AlZnNiZr-p, and ZnNiZr-p were obtained by thermogravimetric analysis (TGA) as reported in Fig. 1. The high temperature calcination induces the transformation of the precursor into homogeneous mixed oxides. In the sample obtained by coprecipitation method, a mass loss between 40 and 240 °C, referring to AlZnNi-c and AlZnNiZr-c, respectively, was attributed to the removal of the interstitial water molecules [21]. The second mass loss between 240 and 450 °C was related to the removal of interlayer carbonate and to the dehydroxylation process [22]. Finally, the loss between 450 and 650 °C was associated to the process of decomposition of the remaining hydroxyls. The loss of structural water occurred between 40 and 450 °C in the ZnNiZr-c precursor. Similarly, a loss was detected between 240 and 450 °C attributed to the removal of carbonate ions. The oxide phases were formed at 450 °C with some remaining hydroxyls adsorbed on their surfaces (see Fig. 1a). The thermogravimetric profiles of the precursors obtained by the modified polymeric method are illustrated in Fig. 1b. From the results, it was found that AlZnNi-p and AlZnNiZr-p presented two mass losses at 406 °C and 446 °C, whereas the precursor containing ZnNiZr-p presented losses at 389 °C and 424 °C, related to the carbon output. It is noteworthy that the precursors were obtained after a pretreatment at 300 °C; therefore, it can be concluded that the second loss is related to the loss of organic matter.
 profiles of (a) AlZnNi-c, AlZnNiZr-c, and ZnNiZr-c; (b) AlZnNi-p, AlZnNiZr-p, and ZnNiZr-p.")
X-ray diffraction patterns of the AZN-c, AZNZ-c, ZNZ-c. AZN-p, AZNZ-p, ZNZ-p, (ZnO), nickel (NiO) and zirconium (ZrO2) oxides are illustrated in Fig. 2. Binary oxides were synthesized by two methods to obtain binary oxide patterns. Particularly, diffraction pattern of AZN-c (see Fig. 2a) presented peaks attributed to NiO (α-JCPDS # 01089-713) and ZnO (β-JCPDS # 010800-075). The NiO phase has cubic symmetry (space group Fm-3m (# 225), while the ZnO phase has hexagonal symmetry belonging to the space group P63mc – # 186). On the other hand, the aluminum oxide (Al2O3) phase was not identified in the samples diffraction patterns (AZN-c and AZNZ-c), indicating the occurrence of an amorphous state. Furthermore, it is reported in the literature that the Al2O3 phase can form a solid solution with ZnO. In the AZN-c and ZNZ-c samples, the high-intensity peak centered at 2θ ≈ 34° is assigned to the ZnO phase. In addition, the ZrO2 phase (cubic and tetragonal crystalline structure) was evidenced in the AZNZ-c samples, but with lower intensity. Finally, the ZNZ-c sample presented the ZrO2 phase with the most visible peaks in relation to the AZNZ-c sample at 2θ ≈ 60°. Concerning the mixed metal oxide obtained by the polymer precursor method, more intense and defined peaks are observed, mainly in AZN-p and AZNZ-p samples. In the case of AZN-p, the peaks occurred around 2θ ~ 43° and 47°, which are relative to the NiO and ZnO phases, respectively. The profile of AZNZ-p presented peaks in 2θ ~ 43°, 47°, 56°, 63° and 67°. In the ZNZ-p sample, ZnO, NiO, and ZrO2 phases were observed.
 XRD patterns of (a) AZN-c, AZNZ-c, and ZNZ-c; (b) AZN-p, AZNZ-p, and ZNZ-p. The phases identified were: ZnO (α), NiO (β), and ZrO2 (λ).")
The reduction peaks contain information related to the metal/support interactions and to the oxidation state, which can be both associated to the displacement of such peaks. The occurrence of reduction peaks at low temperatures can mean a weak interaction of the active phase with the support. On the other hand, if the peaks of reduction occur at high temperatures, this fact may indicate the reduction of the active phase strongly linked to the support or the existence of ternary phases. Therefore, the interaction of the active phase (NiO) with the support can be estimated by observing the peak of reduction through the programmed reduction temperature technique. The reduction profiles for the samples are shown in Fig. 3.
 AZN-c, AZNZ-c, and ZNZ-c; (b) AZN-p, AZNZ-p, and ZNZ-p.")
Analyzing Fig. 3a, it was observed that the AZN-c catalyst exhibited a peak at 549 °C associated to the reduction of the NiO phase weakly bound to the support. This factor is supported by results from the literature, since the Ni interacting with Al2O3 in a tetrahedral coordination is not reduced to temperatures lower than 400 °C [23]. In contrast, the second peak of high intensity occurs at 637 °C, referring to a possible intermediate interaction with other present phases. Another peak (low intensity) at 719 °C was attributed to the stronger interaction of the NiO phase. For the AZNZ-c and ZNZ-c samples, it can be stated that NiO has a low interaction with other present phases. This conclusion is based on the displacement of the peaks at lower temperatures when compared to AZN-c. For the AZNZ-c sample, three reduction peaks were observed at 510, 579 and 673 °C, while for ZNZ-c sample only two reduction peaks were observed at 549 °C and 664 °C. In general, the NiO reduction is summarized by the expression NiO + H2 → Ni° + H2O. Regarding the sample obtained by the polymer precursor method (Fig. 3b), it is noted that the reduction peaks showed few differences. From the results, it can be observed that in the AZN-p sample a displacement to higher temperatures (623 and 691 °C) occurred. Another observed peak was around 567 °C, attributed to a weak interaction with other phases. The AZNZ-p sample presented a displacement for higher temperatures, namely 556, 614, 677 and 679 °C. Finally, the ZNZ-p sample also showed displacements at higher angles and three reduction peaks at 577, 665, and 728 °C, which were attributed to the weak, moderate and strong interactions, respectively.
In order to complement the catalyst reduction studies, the programmed temperature reduction analysis was carried out, in order to observe the behavior of the NiO reduction peak without the presence of other phases. From the graph it is possible to observe a reduction peak around 450 °C; this result demonstrates that the mixed oxides promotes an increase in the interactions of the NiO phase (see Fig. 3c).
The results concerning CO2 desorption are reported in Fig. 4. The AZN-c catalyst showed high-intensity desorption peaks at 66 and 96 °C, in addition to low-intensity peaks around 144 °C and 202 °C, which represent strong basic sites. The AZNZ-c catalyst exhibited peaks at about 74, 101, 131, 176, and 254 °C. Also in Fig. 4c, the ZNZ-c sample showed high-intensity peaks at 79 °C and 100 °C, as well as low-intensity peaks at 128, 188, and 158 °C. Based on the results presented, the samples showed weak basic sites, attributed to the presence of hydroxyl adsorbed on the surfaces of the compounds, forming bicarbonate species (HCO3−). In the samples obtained at 550 °C, no peaks were observed at high temperatures; thus, it was concluded that there are no strong basic sites in these oxides.
 AZN-c, AZNZ-c, and ZNZ-c; (b) AZN-p, AZNZ-p, and ZNZ-p.")
The samples obtained by modified polymer precursor method showed a different behavior (Fig. 4b). The AZN-p showed peaks at 63, 87, 123 and 183 °C, besides peaks at 438 °C and 482 °C. Peaks at low temperatures represent weak basic sites, while peaks at higher temperatures represent stronger basic sites. The AZNZ-p sample had a behavior similar to the AZN-p one, presenting only weak basic sites (70, 89, 113, 142, and 188 °C). Finally, the ZNZ-p sample presented desorption peaks at lower temperatures, around 71, 90, 111, 170, and 205 °C.
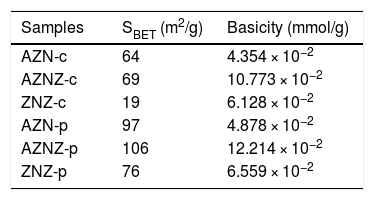
In Table 3 it can be seen that the modified polymeric precursor method clearly influences the number of basic sites, since the samples obtained by modified polymeric precursor showed higher values of basic sites (4.878 × 10−2, 12.214 × 10−2, and 6.559 × 10−2 mmol/g) in relation to the samples obtained by the coprecipitation method (4.354 × 10−2, 10.773 × 10−2, and 6.128 × 10−2 mmol/g). By carrying out the process of mixing the polymers and, consequently, their burning, it is possible to generate a greater amount of basic sites.
Specific surface area (SBET) and number of basic sites of the AZN-c, AZNZ-c, ZNZ-c, AZN-p, AZNZ-p, and ZNZ-p samples.
| Samples | SBET (m2/g) | Basicity (mmol/g) |
|---|---|---|
| AZN-c | 64 | 4.354 × 10−2 |
| AZNZ-c | 69 | 10.773 × 10−2 |
| ZNZ-c | 19 | 6.128 × 10−2 |
| AZN-p | 97 | 4.878 × 10−2 |
| AZNZ-p | 106 | 12.214 × 10−2 |
| ZNZ-p | 76 | 6.559 × 10−2 |
The surface area values of the samples are summarized in Table 3. The samples obtained by the modified polymeric precursor showed the highest values of the surface area. The surface area obtained by AZN-c was around 64 m2/g. Nevertheless, an increase of the surface area occurred when partially replacing Al3+ by Zr4+, thus obtaining a value of 69 m2/g for the catalyst AZNZ-c. The results obtained by the thermogravimetric curves could explain the behavior of this result, since the ZNZ-c sample presented a greater mass loss attributed to carbonate removal and dehydroxylation.
In relation to the samples surface area obtained by the polymeric precursor method, it was verified that a similar behavior occurred with the samples obtained by the chemical coprecipitation method. Moreover, it is noted that there is a significant increase in the surface area of all samples. Analyzing the group, it was found that the AZNZ-p sample exhibited the highest surface area, around 106 m2/g, whereas AZN-p and ZNZ- presented values of 97 and 76 m2/g, respectively (see Table 3).
N2 physisorption isotherms were obtained only for the samples produced by the coprecipitation method. The isotherms for the AZN-p, AZNZ-p and ZNZ-p catalysts are reported in Fig. 5a. All samples showed the hysteresis phenomenon, showing the adsorption (I) and desorption curve at 77 K. These results showed a characteristic of mesoporosity (2–50 nm), in which the condensed gas in the pores does not evaporate easily and, on the contrary, it is recondensed due to the formation of the meniscus in the capillaries. It can be observed that the isotherms presented are not initiated at zero pressure, so it is not possible to verify the inflection point, which corresponds to the formation of the first adsorbed layer that covers the entire surface of the material. In addition, it was not possible to observe the region above a relative pressure (p/p0) of 0.97, which represents the region of large pores [24]. According to the classification established by IUPAC, it was observed that the samples presented a hysteresis type IV behavior. Several factors can be associated to the formation of different isotherms, such as pore forms and the interaction forces between adsorbent and adsorbate [25]. The isotherm presented for AZN-p exhibited a close spacing between adsorption (I) and desorption (II), and its hysteresis cycle occurred in the range of (p/p0) 0.977 to 0.403, related to capillary condensation in the mesopores. The AZNZ-p sample exhibited a behavior similar to the AZN-p one; however, its hysteresis cycle closed at a relative pressure (p/p0) of 0.220, thus demonstrating a larger hysteresis area. In addition, it presented a greater distance between the adsorption (I) and desorption (II) curves. On the other hand, the ZNZ-p presented a differentiated behavior, as its hysteresis cycle closed at a relative pressure of 0.286. From this, it was finally concluded that the AZNZ-p and ZNZ-p samples showed larger pores, whereas AZN-p presented pores of smaller radius. When classifying the hysteresis, AZN-p and AZNZ-p samples presented H3 type hysteresis. This hysteresis type is associated with the presence of particles aggregates and agglomerates that generate pores in the form of slits, with variable sizes and shapes. The ZNZ-p sample presented a H1 hysteresis loop, with the formation of aggregates and agglomerates of particles with variable sizes and shapes [25].
 Pore size distribution of AZN-p, AZNZ-p, and ZNZ-p.")
The pore size distribution curve (dV/dlog(D) vs. pore radius) is shown in Fig. 5b. Initially, peaks in the range of 3–70 nm were noted, suggesting a predominance of mesopores in all samples. The curves for the AZN-p, AZNZ-p and ZNZ-p catalysts indicated a pore concentration with diameters below 15, 10 and 10 nm, respectively. This suggests that essentially all mesoporosity in these samples occurred in small mesopores. The diameters corresponding to the maximum volume of the AZN-p, AZNZ-p and ZNZ-p catalysts were around 38, 27 and 16 nm, respectively. In addition, the presence of pores with diameters greater than 50 nm was noted in all samples.
4ConclusionsModified polymeric precursor and coprecipitation methods were effective in obtaining the mixed metal oxides (NiO, ZnO, Al2O3, and ZrO2). Furthermore, it was found that the synthesis method influenced mainly on the surface area. The samples obtained by the polymer precursor method presented higher surface area values, which has allowed to conclude that the decomposition of the polymers provides a higher porosity, hence a greater surface area and a larger amount of basic sites. Another factor to be emphasized is that the polymeric precursor method favored stronger interactions between NiO and the other present phases, which reflected in obtaining peaks of reduction at higher temperatures. Therefore, it is concluded that the application of the methods studied in this article depends mainly on the application of the mixed oxide, since each method has its particularity in relation to structural properties.

