


De entre as síndromes poliglandulares autoimunes (SPGA), a mais frequente é a SPGA tipo 2, que designa a associação de doença de Addison (DA) autoimune com diabetes mellitus (DM) tipo 1 e/ou disfunção tiroideia autoimune. Estas patologias major estão presentes com diversas combinações entre si, podendo ainda coexistir com outras doenças autoimunes minor, endócrinas ou não. A SPGA tipo 2 é mais frequente em mulheres adultas, com prevalência de 2 casos por cada 100.000 habitantes.
A síndrome de Carpenter (designação que tem vindo a cair em desuso) é um caso particular de SPGA tipo 2, correspondendo a cerca de 10% do total, em que estão presentes todas as manifestações major (DA, DM tipo 1 e disfunção tiroideia autoimune).
Os autores apresentam um caso clínico de um jovem do sexo masculino, com diagnóstico de DM tipo 1 desde a infância, que foi internado para estudo de quadro de vómitos, dor abdominal e alterações hidroeletrolíticas (hiponatrémia grave e hipercaliémia) e cuja investigação etiológica culminou no diagnóstico de SPGA tipo 2.
O tratamento desta síndrome implica as mesmas medidas utilizadas para terapêutica das patologias diagnosticadas isoladamente, embora por vezes sejam necessários pequenos ajustamentos.
Com este caso pretende-se promover o debate para eventualmente instituir protocolos de diagnóstico e seguimento de doentes com manifestações de autoimunidade, para o seu atempado diagnóstico e melhoria na qualidade de vida e sobrevida.
Among the entire range of autoimmune polyglandular syndromes (APS), the most frequent is APS type 2, which designates the association between autoimmune Addison's disease (AD) and Diabetes mellitus (DM) type 1 and/or autoimmune thyroid malfunction. These major diseases can present themselves in different combinations, and sometimes they may also coexist with other minor autoimmune diseases, endocrinological or not. APS type 2 is more frequent in adult women, with a prevalence of 2 cases per 100.000 inhabitants.
Carpenter's syndrome (although this designation is being abandoned) designates a particular case of APS type 2 – about 10% of total APS – in which all of the major diseases are present (AD, DM type 1 and autoimmune thyroid malfunction).
The authors present a clinical case of a young man, who had been diagnosed DM type 1 during infancy, that was admitted to the infirmary for etiological study of vomiting, abdominal pain and hydroeletrolyte disturbance (severe hyponatremia and hyperkalemia). This study led to a Carpenter syndrome diagnosis.
Carpenter syndrome treatment involves the same approach as if the pathologies had been diagnosed by themselves, although sometimes small adjustments should be made.
With this clinical case report we wish to promote the debate toward establishing diagnosis protocols e follow-up procedures in patients with autoimmune diseases, aiming its attempted diagnosis and quality of life improval.
Estão descritas desde longa data associações entre diversas doenças autoimunes (endócrinas e não endócrinas), que são denominadas por síndrome poliglandular autoimune (SPGA). Existem múltiplas combinações destas patologias autoimunes, que surgem com frequências variáveis.
A SPGA tipo 2 é a forma mais comum de SPGA. É, no entanto, uma doença rara, com uma prevalência de 1,4-2 casos por 100.000 habitantes1, que afeta sobretudo mulheres adultas (20-40 anos). Embora seja uma doença poligénica, associa-se sobretudo à presença de antigénios HLA DR3 e HLA DR4, com padrão de transmissão aparentemente autossómico dominante com penetrância incompleta1.
O diagnóstico é estabelecido quando se verifica a presença no mesmo doente de doença de Addison autoimune (presente em 100% dos casos, por definição2), diabetes mellitus tipo 1 (DM tipo 1) autoimune e/ou doença tiroideia autoimune (síndrome de Schmidt). Podem coexistir com outras manifestações menos frequentes (4-11% dos casos) como vitiligo, gastrite autoimune, hipogonadismo hipergonadotrófico, hepatite autoimune e alopecia, assim como com outras condições raras associadas à doença (frequência inferior a 1%), nomeadamente Miastenia gravis, artrite reumatoide e síndrome de Sjogren3.
A primeira manifestação de doença autoimune é habitualmente DM tipo 1, seguida pelo aparecimento das outras (cuja ordem cronológica é variável4). Apenas 10% dos doentes apresentam a tríade completa das manifestações major5 (também denominada síndrome de Carpenter6, designação em utilização cada vez menos frequente).
O tratamento da síndrome envolve as medidas terapêuticas que são utilizadas nestas patologias quando surgem isoladamente4,5.
O prognóstico dos indivíduos com SPGA tipo 2 relaciona-se sobretudo com as manifestações de insuficiência suprarrenal, responsáveis pela morbilidade e mortalidade destes doentes5.
Caso clínicoOs autores apresentam o caso de um jovem do sexo masculino de 23 anos, de raça caucasiana, seguido em consulta de endocrinologia desde o diagnóstico de DM tipo 1 na infância (medicado habitualmente com insulina isofânica 18+0+16U), que recorreu ao serviço de urgência por quadro de dor abdominal difusa, vómitos e hiperglicemia (glicemia capilar de 338mg/dL).
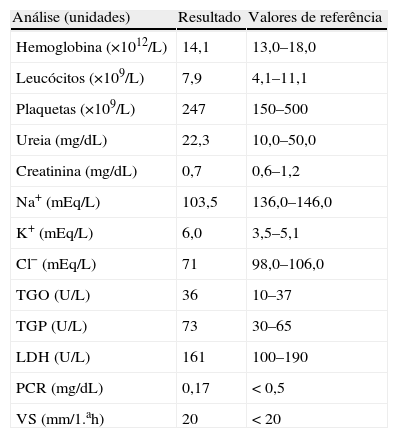
Após observação inicial foram pedidos exames analíticos, que revelavam hiponatrémia grave (103mEq/dL) e hipercaliémia (6,0mEq/dL) (ver tabela 1 com restante estudo analítico do SU). Colocada hipótese diagnóstica de se tratar de cetoacidose diabética, que foi rapidamente excluída com a ausência de cetonemia e cetonúria, assim como pela gasometria arterial (sem alterações do equilíbrio ácido-base). Para compensação clínica e estudo etiológico do quadro clínico foi internado, após correção de hipercaliémia com administração de 8U insulina regular (também para melhor controlo glicémico), salbutamol nebulizado e furosemida e.v.
Resultados analíticos à admissão no serviço de urgência
| Análise (unidades) | Resultado | Valores de referência |
| Hemoglobina (×1012/L) | 14,1 | 13,0–18,0 |
| Leucócitos (×109/L) | 7,9 | 4,1–11,1 |
| Plaquetas (×109/L) | 247 | 150–500 |
| Ureia (mg/dL) | 22,3 | 10,0–50,0 |
| Creatinina (mg/dL) | 0,7 | 0,6–1,2 |
| Na+ (mEq/L) | 103,5 | 136,0–146,0 |
| K+ (mEq/L) | 6,0 | 3,5–5,1 |
| Cl− (mEq/L) | 71 | 98,0–106,0 |
| TGO (U/L) | 36 | 10–37 |
| TGP (U/L) | 73 | 30–65 |
| LDH (U/L) | 161 | 100–190 |
| PCR (mg/dL) | 0,17 | <0,5 |
| VS (mm/1.ah) | 20 | <20 |
Cl−: cloro; K+: potássio; LDH: desidrogenase láctea; Na+: sódio; PCR: proteína C reativa; TGO (AST): aspartato aminotransferase; TGP (ALT): alanina aminotransferase; VS: velocidade de sedimentação.
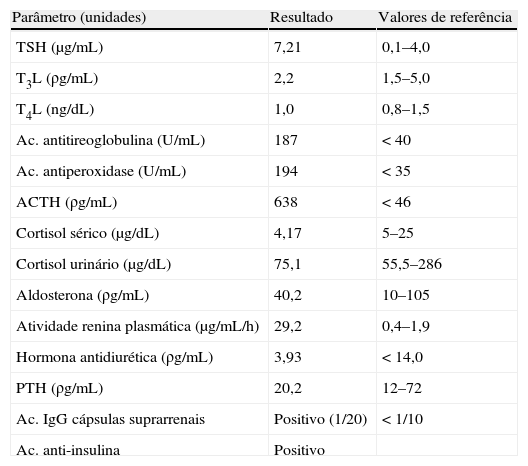
Já no internamento foram pedidos doseamentos de ACTH, cortisol plasmático e urinário pela suspeita de insuficiência suprarrenal (apesar da ausência de outras características clínicas desta entidade, incluindo hiperpigmentação cutânea). Dada a dificuldade de controlo metabólico e hidroeletrolítico (hiperglicemias mantidas, hiponatrémia de difícil correção3, poliúria superior a 8.000cc/24h), foi requisitado doseamento de aldosterona sérica e atividade plasmática de renina.
Foram requisitados exames de imagem, todos sem alterações (TC crânio-encefálica, RM da hipófise e TC das glândulas suprarrenais).
Cerca de 4 dias após admissão na enfermaria, iniciou empiricamente substituição glicocorticoide com hidrocortisona 15mg/dia (10+5) com boa resposta a nível de correção de ionograma. Para a alta clínica tentou-se medicação somente com prednisolona, tendo havido agravamento da hiponatrémia, pelo que foi então associada fludrocortisona 0,5mg id.
Com o estudo efetuado compatível com insuficiência suprarrenal, com disfunção tiroideia subclínica e presença de autoanticorpos antitiroideus (ac. antitireoglobulina e ac. antiperoxidase), colocou-se hipótese diagnóstica de SPGA tipo 2, que foi confirmada com a positividade de anticorpos dirigidos contra as cápsulas suprarrenais (resultados completos na tabela 2).
Resultados do estudo de autoimunidade e eixo hipófise-suprarrenal
| Parâmetro (unidades) | Resultado | Valores de referência |
| TSH (μg/mL) | 7,21 | 0,1–4,0 |
| T3L (ρg/mL) | 2,2 | 1,5–5,0 |
| T4L (ng/dL) | 1,0 | 0,8–1,5 |
| Ac. antitireoglobulina (U/mL) | 187 | <40 |
| Ac. antiperoxidase (U/mL) | 194 | <35 |
| ACTH (ρg/mL) | 638 | <46 |
| Cortisol sérico (μg/dL) | 4,17 | 5–25 |
| Cortisol urinário (μg/dL) | 75,1 | 55,5–286 |
| Aldosterona (ρg/mL) | 40,2 | 10–105 |
| Atividade renina plasmática (μg/mL/h) | 29,2 | 0,4–1,9 |
| Hormona antidiurética (ρg/mL) | 3,93 | <14,0 |
| PTH (ρg/mL) | 20,2 | 12–72 |
| Ac. IgG cápsulas suprarrenais | Positivo (1/20) | <1/10 |
| Ac. anti-insulina | Positivo |
Ac.: anticorpos; ACTH: hormona adrenocorticotrófica; PTH: paratormona; TSH: hormona estimulante da tiroide; T3L: triiodotironina livre; T4L: tiroxina livre.
O doente teve alta do internamento medicado com prednisolona 10+5mg id, fludrocortisona 0,1mg id, levotiroxina 50ug id, insulina glargina 0+0+30U e insulina humana 10+10+10U, mantendo seguimento na consulta de endocrinologia e encontrando-se assintomático (controlo analítico revela ionograma estabilizado, HbA1c de 7,4 e TSH ainda de 5,54 com T4L 1,01, mantendo terapêutica com levotiroxina).
DiscussãoA SPGA tipo 2 é a forma mais comum de SPGA, embora seja uma patologia rara. Estes doentes apresentam uma combinação de doença de Addison autoimune, DM tipo 1 e disfunção tiroideia autoimune, sendo pouco comum a associação destas 3 entidades no mesmo doente. A presença predominante de SPGA tipo 2 em indivíduos do sexo feminino relaciona-se com a maior frequência de disfunção tiroideia neste sexo, que está presente em 90% dos doentes.
Na presença de doença de Addison autoimune ou DM tipo 1, não é consensual a indicação para pesquisa de existência de outras entidades incluídas na SPGA tipo 2 (tendo presente a possibilidade de existência de marcadores específicos, mesmo que ainda sem doença clinicamente detetável)7–9. Deste modo, seria possível diagnóstico mais precoce e início de terapêutica dirigida atempada, com consequente diminuição de morbi e mortalidade associada10. Quando se opta pela realização do referido estudo, recomenda-se a avaliação não só de autoimunidade e disfunção suprarrenal, pancreática e tiroideia, mas também gonadal, gástrica e intestinal5 (pela possibilidade de ocorrência de hipogonadismo, gastrite autoimune e doença celíaca, respetivamente).
Por se tratar de entidade com padrão de transmissão familiar, devem também ser pesquisados os familiares em primeiro grau destes doentes5,7.
O tratamento destes doentes apresenta algumas particularidades, apesar de os princípios gerais serem idênticos aos utilizados na abordagem às doenças diagnosticadas isoladamente.
A crise suprarrenal é uma emergência médica, cujo tratamento deve ser iniciado empiricamente ainda antes da confirmação diagnóstica, com administração de fluidoterapia agressiva e corticoterapia com hidrocortisona 100mg 6/6h, assim como outras medidas de suporte entretanto necessárias (como o suporte vasopressor, por exemplo).
O tratamento da hiponatrémia no doente estável e sem sintomatologia neurológica deve obedecer a um ritmo de correção não superior a 0,5mmol/h ou 12mmol/24h, pelo risco de desmielinização osmótica. Se o doente apresenta sintomatologia neurológica devida a hiponatrémia (como na presença de convulsões) ou outra lesão cerebral (por exemplo tumor ou meningite), o ritmo de correção deverá ser de 1,5-2mmol/h nas primeiras 3-4h ou até melhoria dos sintomas, mantendo o objetivo de não ultrapassar os 12mmol nas 24h.
Para tratamento crónico de insuficiência suprarrenal deve-se tentar utilizar padrão de administração de fármacos que se assemelhe ao da libertação endógena11. O fármaco de eleição para reposição glicocorticoide é a hidrocortisona (15-25mg diários na maioria dos doentes, devendo ser administrada metade a 2 terços da dose ao acordar e o restante ao início da tarde1), podendo contudo ser utilizados glicocorticoides de ação prolongada (prednisolona e dexametasona). A substituição mineralocorticoide é assegurada com fludrocortisona, cuja dose terapêutica habitual é de 0,1mg diários. Nos doentes do sexo feminino é necessária ainda suplementação para substituição androgénica, o que é feito com DHEA (habitualmente 25-50mg).
A manutenção do controlo glicémico é feita, tal como nos restantes doentes diabéticos tipo 1, com insulinoterapia otimizada. É necessária particular atenção ao ajuste de dose de insulina, em relação com a patologia suprarrenal (diminuição da dose nos estados de insuficiência não corrigida e aumento de dosagens quando é necessária reposição glicocorticoide em doses elevadas).
No tratamento do doente com disfunção tiroideia é necessário ter algumas precauções especiais. O início recente de terapêutica com levotiroxina, pela elevação de depuração hepática de cortisol, pode precipitar insuficiência suprarrenal em indivíduos com esta doença subclínica. De igual modo, o aparecimento de hipertiroidismo em doente com doença de Addison implica aumento de suplementação glicocorticoide até atingimento de estado eutiroideu.
ConclusãoCom este caso clínico, os autores pretendem chamar a atenção para as várias possibilidades diagnósticas a colocar em cada caso clínico, mesmo quando estas são raras. Com efeito, um jovem do sexo masculino com uma apresentação clínica que poderia ser enquadrada em diversas patologias (gastroenterite, cetoacidose diabética, hipertiroidismo, …) veio a revelar ser portador de uma doença rara na população geral.
Pretende-se ainda salientar a importância/necessidade de estabelecer protocolos de diagnóstico e seguimento consensuais de diversas patologias autoimunes, com possibilidade de melhoria na qualidade de vida e sobrevida destes doentes.
Conflito de interessesOs autores declaram não haver conflito de interesses.
seguindo a fórmula Vinfundir=[Δ desejada Na+×(água corporal+1)]/(Na+doente−Na+soro infundido). O volume total de soro a infundir é dado por esta fórmula, em que é multiplicada a variação de concentração de sódio desejada pela (água corporal total+1) e este resultado é dividido pela diferença entre o sódio sérico do doente e a concentração de sódio infundido (por exemplo, 1000ml de soro fisiológico correspondem a 154mmol/L de sódio).
