





Os feocromocitomas e paragangliomas são tumores raros com origem nas células derivadas da crista neural. Os primeiros têm localização na medula da suprarrenal e os últimos no sistema nervoso simpático e parassimpático. As suas manifestações clínicas dependem da localização, do perfil secretor e do potencial maligno do tumor. Alguns surgem no contexto de síndromes familiares, outros esporadicamente. Os recentes avanços da genética têm demonstrado que cerca de 1/3 desses tumores tem origem familiar. Isso significa que pelo menos alguns doentes com tumores aparentemente esporádicos (e respetivos familiares) beneficiam da identificação da mutação responsável pela doença, nomeadamente quando se trata de doentes jovens, com feocromocitoma maligno, multifocal ou bilateral ou com paragangliomas. Assim, poder‐se‐á fazer uma vigilância mais adequada, diagnósticos mais precoces e terapêuticas atempadas.
Este artigo pretende focar as características dos tumores provocados pelas mutações até agora identificadas, bem como considerar aspetos a ter em conta perante a possibilidade de pedir estudo genético em doentes com feocromocitoma ou paraganglioma aparentemente esporádico.
Pheochromocitoma and paraganglioma are rare tumors with origin in the cells derived from the neural crest. The former are adrenal medulla tumors and the later are tumors of sympathetic and parasympathetic nervous system. Their clinical presentation depends on its location, secretory profile and malignant potential. Some of them belong to familiar syndromes others arise sporadically. Recent advances in genetics have shown that about one third of these tumors are familiar. This means that some patients with apparently sporadic tumors and their families will benefit with the identification of the mutated gene, particularly those who are young, those who have malignant, bilateral or multifocal pheochromocytoma or paragangliomas. This could help us to do a better surveillance, an earlier diagnosis and timely treatment of the affected patients.
This article aims to address the characteristics of tumors due to the mutations identified so far, focusing on the information that can help us in the decision of performing a genetic test in patients with apparently sporadic pheochromocytoma or paraganglioma.
Os feocromocitomas (FEO) e paragangliomas (PGL) são tumores com origem em células derivadas da crista neural. São muitas as classificações existentes, mas segundo a da Organização Mundial de Saúde (OMS) FEO é um tumor situado na medula da glândula suprarrenal, enquanto PGL é um tumor com localização extra‐adrenal: no sistema nervoso parassimpático (em células não cromafins, ao longo dos nervos cranianos), habitualmente na cabeça e pescoço, sendo que apenas 5% são funcionantes, ou no sistema nervoso simpático (em células cromafins do tórax, abdómen ou pélvis), secretando em regra catecolaminas1,2. É a classificação da OMS que vai ser utilizada ao longo do texto.
Estes tumores são raros, com uma incidência entre 2‐8 casos por milhão por ano3, sendo maior entre os 50‐60 anos1, não existindo diferenças entre os sexos4. Até 1999 acreditava‐se que apenas cerca de 10% destes tumores eram familiares5. Desde então, muitos estudos têm vindo a ser feitos, com o objetivo de compreender melhor a genética dos mesmos e, graças aos resultados obtidos, acredita‐se que cerca de 1/3 tenham uma origem familiar3. Perante este facto, é necessário repensar as estratégias de estudo dos FEO e PGL, tendo em conta que isso pode ter implicações no tratamento, prognóstico e vigilância dos doentes e dos seus familiares3.
Síndromes familiares de feocromocitomas e paragangliomasCerca de 10% dos doentes com FEO ou PGL têm história familiar da doença, ou seja, têm formas sindrómicas com história familiar positiva3. Até cerca de 30% dos doentes com formas aparentemente esporádicas da doença apresentam mutações herdadas dos seus progenitores3. Isto significa que existem formas genéticas que podem passar despercebidas, devendo‐se isso a vários fatores: algumas mutações identificadas têm imprinting materno (o gene fica inativo ao passar pelo progenitor feminino), outras têm penetrância incompleta ou expressão variável e ainda podem surgir mutações de novo6.
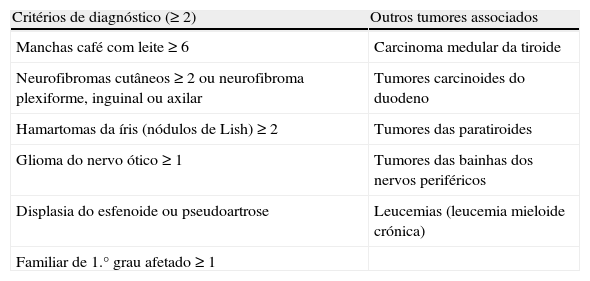
Neurofibromatose tipo 1A neurofibromatose tipo 1 (NF1) ou doença de Von Recklinghausen é uma doença autossómica dominante que ocorre em 1:3.000 indivíduos4,7. Deve‐se a mutações inativadoras no gene da neurofibromina, um supressor tumoral, localizado no cromossoma 17q11.26,7. Caracteriza‐se pela presença de múltiplos tumores (critérios de diagnóstico na tabela 1), entre os quais FEO/PGL, que podem surgir em 0,1‐5,7% dos casos6–8. Se considerarmos os doentes com NF1 hipertensos, essa percentagem ascende aos 50%6–8.
Características clínicas da neurofibromatose tipo 1
| Critérios de diagnóstico (≥2) | Outros tumores associados |
| Manchas café com leite ≥6 | Carcinoma medular da tiroide |
| Neurofibromas cutâneos ≥2 ou neurofibroma plexiforme, inguinal ou axilar | Tumores carcinoides do duodeno |
| Hamartomas da íris (nódulos de Lish) ≥2 | Tumores das paratiroides |
| Glioma do nervo ótico ≥1 | Tumores das bainhas dos nervos periféricos |
| Displasia do esfenoide ou pseudoartrose | Leucemias (leucemia mieloide crónica) |
| Familiar de 1.° grau afetado ≥1 |
Nesta síndrome, o diagnóstico de FEO/PGL é feito na quinta década de vida (idade média: 42 anos), a mesma que na população em geral6,7, assemelhando‐se assim aos casos esporádicos na forma de apresentação7. Na maioria dos casos os tumores são FEO benignos e unilaterais (84%), seguidos de FEO bilaterais (10%), sendo os PGL do sistema nervoso simpático os mais raros (6%)6–8. Os casos de malignidade surgem em cerca de 12%, coincidindo também com a população em geral6. Os FEO/PGL funcionantes da NF1 produzem sobretudo noradrenalina, pelo que a hipertensão e os sintomas adrenérgicos dominam o quadro clínico6–8. Apesar disso, 22% dos casos surgem sem esses sintomas7. Uma vez que os doentes apresentam desde cedo as típicas manchas café com leite na pele, o diagnóstico é pouco provável em casos de FEO/PGL esporádicos6,7.
As mutações que originam esta doença são habitualmente de novo, o que dificulta o estudo genético. No entanto, as suas características fenotípicas (tabela 1) tornam‐no dispensável3.
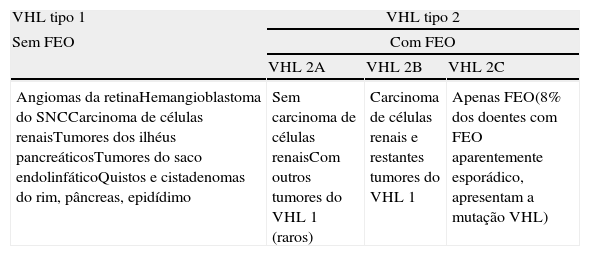
Doença de Von Hippel‐LindauA doença de Von Hippel‐Lindau (VHL) é uma doença autossómica dominante, com expressão variável e penetrância relacionada com a idade (>90% aos 60 anos)3. Tem uma incidência de 1:36.000 nascimentos7. É causada por mutações no gene supressor tumoral VHL, localizado no cromossoma 3p25.56, que codifica a proteína pVHL envolvida na angiogénese, através da regulação da atividade do fator 1 alfa induzido pela hipoxia (HIF‐1α)6,7. A perda de função desta proteína leva à manifestação clínica da doença, caracterizada pela presença de tumores benignos e malignos (tabela 2)6. Já foram identificadas mais de 300 mutações neste gene e 20% das famílias apresentam mutações de novo6. Assim, é uma das mutações prováveis nos casos de FEO/PGL aparentemente esporádicos6.
Características clínicas da doença de Von Hippel‐Lindau
| VHL tipo 1 | VHL tipo 2 | ||
| Sem FEO | Com FEO | ||
| VHL 2A | VHL 2B | VHL 2C | |
| Angiomas da retinaHemangioblastoma do SNCCarcinoma de células renaisTumores dos ilhéus pancreáticosTumores do saco endolinfáticoQuistos e cistadenomas do rim, pâncreas, epidídimo | Sem carcinoma de células renaisCom outros tumores do VHL 1 (raros) | Carcinoma de células renais e restantes tumores do VHL 1 | Apenas FEO(8% dos doentes com FEO aparentemente esporádico, apresentam a mutação VHL) |
Os FEO da doença de VHL surgem em cerca de 10‐20% dos doentes, com idade média de 30 anos6. Os doentes com VHL tipo 1 apresentam perda de função da pVHL devido a deleções do seu gene, desenvolvendo hemangioblastomas do sistema nervoso central ou da retina, bem como carcinoma de células renais, mas não apresentam risco de FEO (tabela 2)8. Os doentes com VHL tipo 2 têm mutações específicas missense do gene VHL, podendo desenvolver hemangioblastomas e FEO, com elevado (tipo 2 A) ou baixo (tipo 2B) risco de carcinoma de células renais8. Uma pequena percentagem de doentes (tipo 2C) desenvolvem apenas FEO8. Neste tipo de doença as mutações missense que ocorrem não acarretam perda de função da pVHL de forma significativa, pelo que os outros tumores típicos da doença não se manifestam, mesmo em casos de homozigotia8. Assim, a identificação da mutação em causa tem vantagens em termos de prognóstico e seguimento dos doentes e da sua família3,8.
Nesta síndrome, os tumores mais frequentes são os FEO, habitualmente bilaterais e múltiplos (mais de 50%), no entanto, PGL funcionantes e não funcionantes também podem surgir (menos de 10%)6. Menos de 5% dos doentes têm formas malignas6 e habitualmente são PGL do sistema nervoso simpático8. Produzem apenas noradrenalina devido à baixa expressão da feniletanolamina‐N‐metiltransferase (PNMT), pelo que o seu diagnóstico bioquímico se baseia na deteção de elevados níveis de normetanefrina no plasma ou na urina6.
Neoplasias endócrinas múltiplas tipo 1 e tipo 2Neoplasia endócrina múltipla tipo 1 (MEN 1) é uma síndrome autossómica dominante, com uma incidência de 1:30.000 nascimentos7. É causada por mutações que envolvem o gene supressor tumoral, codificador da proteína menina, localizado no cromossoma 11q13. É caracterizada por hiperparatiroidismo primário, que surge em 95% dos doentes afetados, associado a tumores da hipófise (em 30‐40% dos doentes, sendo o prolactinoma o mais frequente) e do pâncreas (em 30‐70% dos doentes, sendo o gastrinoma o mais habitual)9.
Nesta síndrome, a suprarrenal também pode apresentar patologia, frequentemente através da presença de adenomas não funcionantes do córtex (em 40% dos doentes), surgindo FEO em menos de 1% dos casos9. De facto, apenas foram identificados 10 casos de FEO nesta síndrome, todos unilaterais, raramente malignos, produtores de noradrenalina, manifestando‐se assim com hipertensão7. Em nenhum deles o FEO/PGL foi a primeira manifestação de doença7. Esta não é por isso uma síndrome a considerar nos casos esporádicos7.
Neoplasia endócrina múltipla tipo 2 (MEN 2) é uma doença autossómica dominante causada por mutações no proto‐oncogene RET, localizado no cromossoma 10q11.2, que codifica um recetor transmembranar tirosina cinase envolvido na regulação da proliferação celular e apoptose. Existem 3 subtipos de doença: MEN 2A, em que 95% dos doentes apresenta carcinoma medular da tiroide, 50% apresentam FEO e 15‐30% desenvolvem hiperparatiroidismo primário devido a adenoma ou carcinoma das paratiroides; MEN 2B, em que todos os doentes desenvolvem carcinoma medular da tiroide, 50% apresentam FEO, surgindo estes tumores associados a ganglioneuromas múltiplos das mucosas e a um fenótipo marfanoide; um terceiro subtipo caracterizado por carcinoma medular da tiroide isoladamente6,10. Aproximadamente 90% dos doentes com MEN têm o subtipo 2A e cerca de 85% desses apresenta mutação do codão 634, exão 116. Noventa e cinco por cento dos casos de MEN 2B são devidos a mutação do codão 918, exão 16 do gene RET6. Raros tumores apresentam mutações noutros codões7. Mutações missense no domínio intracelular da tirosina cinase (Met918Thr) estão associadas ao subtipo MEN 2B (a doença manifesta‐se em idade mais precoce que no subtipo MEN 2A)3. Mutações missense no domínio extracelular que causa dimerização anormal e auto‐ativação das proteínas mutantes estão associadas ao subtipo MEN 2A3.
Os tumores produtores de catecolaminas que surgem no MEN 2 são sobretudo FEO, benignos e bilaterais em mais de 50% dos doentes6. Menos de 5% são malignos, sendo o risco maior em crianças com MEN 2B6, e se os tumores tiverem grandes dimensões7. O diagnóstico é feito habitualmente entre os 30‐40 anos6, após o de carcinoma medular da tiroide, pelo que não é de esperar encontrar esta síndrome em casos de FEO/PGL aparentemente esporádicos3. No entanto, detetou‐se mutação no gene RET em cerca de 5% desses tumores3. Apresentam sobre‐expressão da enzima PNMT, que converte a noradrenalina em adrenalina, pelo que o seu diagnóstico bioquímico se baseia na identificação de níveis elevados de adrenalina ou metanefrina no plasma ou urina6,7. Clinicamente, caracterizam‐se por paroxismos de palpitações, ansiedade e cefaleias, mais do que hipertensão6,7.
O complexo succinato desidrogenaseA succinato desidrogenase (SDH) é uma enzima mitocondrial (complexo mitocondrial II), que intervém nos processos de geração de energia (fig. 1). Está ancorada à membrana mitocondrial interna e é constituída por 4 subunidades: A, B, C e D. A SDHA é uma flavoproteína hidrofílica que serve de local de ligação ao substrato e, juntamente com a proteína também hidrofílica SDHB, forma o local catalítico da enzima. As subunidades SDHC e SDHD são hidrofóbicas e constituem a zona de ancoragem à membrana mitocondrial interna, bem como o local de ligação à ubiquinona. Este complexo enzimático é diferente de todos os outros da cadeia respiratória: não é transmembranar, não transfere hidrogeniões para o espaço transmembranar através da membrana mitocondrial interna e intervém no ciclo de Krebs (catalisa a conversão de succinato em fumarato)2,3.
, enzima mitocondrial (complexo mitocondrial II), que está ancorada à membrana mitocondrial interna e é constituída por 4 subunidades: A, B, C e D. Intervém nos processos de geração de energia e é uma das enzimas que intervém nas reações do ciclo de Krebs, catalisando a transformação de succinato (S) em fumarato (F).")
Esquema do complexo succinato desidrogenase (SDH), enzima mitocondrial (complexo mitocondrial II), que está ancorada à membrana mitocondrial interna e é constituída por 4 subunidades: A, B, C e D. Intervém nos processos de geração de energia e é uma das enzimas que intervém nas reações do ciclo de Krebs, catalisando a transformação de succinato (S) em fumarato (F).
Múltiplas mutações que envolvem várias proteínas que constituem o complexo SHD têm vindo a ser associadas a formas familiares de FEO/PGL2–4,11–14. Muito se tem especulado acerca da razão pela qual as referidas mutações causam este tipo de tumores2. Acredita‐se que a via da tumorigénese destas mutações esteja relacionada com a angiogénese2. As diferenças de fenótipo entre as síndromes derivadas das várias mutações também não estão bem esclarecidas – é provável que as mutações que envolvem as subunidades catalíticas acarretem perda total de função, originando assim formas mais agressivas da doença, ao contrário das mutações nas subunidades de ancoragem2.
Síndrome de paragangliomas familiares tipo 1As mutações do gene codificador da subunidade SDHD, localizado no cromossoma 11q23, foram as primeiras a ser identificadas2,7. Têm imprinting materno e penetrância relacionada com a idade (50% aos 31 anos e 80% aos 50)3,4,7. Dão origem a PGL benignos e multifocais da cabeça e do pescoço (cadeias nervosas parassimpáticas), mais frequentemente a nível da bifurcação da carótida e raramente no glomo timpânico2,11,12. Apesar de raros, podem surgir PGL das cadeias simpáticas e FEO2,7, habitualmente bilaterais e multifocais4,11, bem como doença metastática (menos de 5% dos casos)2,4. Por vezes são recorrentes6. O diagnóstico é feito habitualmente na terceira e quarta década de vida (idade média de diagnóstico: 35 anos)3,6,12. Produzem habitualmente noradrenalina4.
Existe uma correlação entre o genótipo e o fenótipo: mutações nonsense e splicing estão associadas a diagnóstico em idades mais precoces e à presença de FEO e PGL da cabeça e pescoço, provavelmente por originarem proteínas truncadas ou ausência da proteína codificada, enquanto que mutações missense dão origem a proteínas ainda com alguma função2.
Síndrome de paragangliomas familiares tipo 2Esta síndrome é muito rara e autossómica dominante6,7. Resulta da mutação do gene localizado no cromossoma 11q13.17, que codifica a proteína SDHAF2, enzima envolvida na flavinação da subunidade SDHA (adição do dinucleótido flavina‐adenina, grupo prostético da subunidade), tornando‐a ativa e permitindo a funcionalidade de todo o complexo SDH2,11. Até à atualidade foram identificadas 2 famílias com esta síndrome, apresentando uma mutação missense, p.Gly78Arg, com imprinting materno e elevada penetrância: dos 42 portadores, 37 já manifestaram a doença2,6,7,11. Todos os doentes, com idades inferiores a 45 anos, têm apenas PGL da cabeça e do pescoço11.
Síndrome de paragangliomas familiares tipo 3Esta síndrome rara é autossómica dominante e deve‐se a mutações no gene que codifica a SDHC, localizado no cromossoma 1.q214,6,7. Não tem imprinting materno6,7. É caracterizado por PGL parassimpáticos da cabeça e pescoço, não funcionantes, benignos (menos de 5% são malignos) e raramente multifocais4,7. PGL funcionantes e FEO são extremamente raros, mas já foram descritos2,3. A idade média de diagnóstico é 38 anos12.
Síndrome de paragangliomas familiares tipo 4A síndrome de PGL familiares tipo 4 deve‐se a mutações inativadoras do gene localizado no cromossoma 1p36.1‐35, que codifica a subunidade SDHB, causando destabilização do complexo SDH e ativando vias que culminam na angiogénese, desencadeadas pela hipoxia7.
Nesta síndrome os tumores são sobretudo malignos: as taxas de malignidade variam entre 34‐70%2,6,8. Um FEO maligno tem uma probabilidade de 50% de ter mutação do gene SDHB2. A maioria dos tumores produz catecolaminas, especialmente noradrenalina4, embora cerca de 10% sejam bioquimicamente silenciosos ou produzam apenas dopamina2. Apesar disso, os sintomas de apresentação não são típicos, o que atrasa o diagnóstico – muitos deles surgem apenas com a metastização ou efeito de massa2,6,14. Em termos globais, os PGL simpáticos secretores malignos são os mais frequentes, seguidos dos FEO e dos PGL benignos da cabeça e pescoço2,4,6,14. Cerca de 2/3 dos doentes apresentam‐se com PGL toracoabdominais e 1/3 tem doença multifocal2. Não há uma relação clara entre o genótipo e o fenótipo, no entanto, há vários fatores que parecem relacionar‐se com pior prognóstico: idade jovem à data do diagnóstico, tumores de grandes dimensões, secretores de dopamina e com mutações missense2. A própria mutação tem valor prognóstico, já que se relaciona com maior mortalidade13.
A idade média de diagnóstico é 30 anos2,6. A penetrância é elevada e relaciona‐se com a idade: 45% aos 40 anos, 77% aos 50 anos2,7,13,14. Assim, perante doença metastática, mesmo com história familiar negativa, a pesquisa da mutação do gene SDHB deve ser uma preocupação6,8.
Esta síndrome parece relacionar‐se com um risco aumentado de carcinoma de células renais, tumores do estroma gastrointestinal (GIST), carcinoma papilar da tiroide e da mama3,6–8.
Mutações rarasApesar de codificar a maior subunidade do complexo SDH, as mutações no gene da SDHA não se têm associado com frequência a síndromes de PGL11. Inicialmente apenas surgia como a mutação subjacente a uma doença neurodegenerativa conhecida como síndrome de Leigh6. No entanto, recentemente foi identificada a primeira mutação (c.1765C>T, p.Arg589Trp – exão 13) num doente com um PGL abdominal secretor de catecolaminas, sem história familiar, estando também presente em 4,5% de um grupo de 202 doentes com FEO e PGL15. A razão para o facto de ser a única subunidade que menos tem estado ligada a síndromes de PGL não é conhecida. Especula‐se que possa estar relacionada com reduzida penetrância ou ao facto dessas mutações induzirem alterações tão graves que sejam incompatíveis com a vida11.
O gene TMEM127 codifica uma proteína transmembranar com 3 domínios que atua como supressor tumoral e localiza‐se no gene 2q11.211. Ainda não é clara a sua função, mas pode relacionar‐se com o transporte de proteínas entre a membrana citoplasmática e alguns organelos, como o complexo de Golgi e os lisossomas, bem como nas vias de sinalização da mTOR11. Mutações neste gene têm estado envolvidos no desenvolvimento de FEO habitualmente bilaterais e benignos, com uma transmissão autossómica dominante3. A idade média de diagnóstico é 41,5 anos, portanto mais tardiamente que nos doentes com mutações dos genes VHL e SDHB3. Nos estudos efetuados, apenas foi identificado em cerca de 2% dos doentes com FEO ou PGL3.
Mutações do gene MAX, supressor tumoral localizado no cromossoma 14q23, também têm vindo a ser identificadas em doentes com FEO e PGL16. Parecem ser responsáveis por 1,12% dos doentes com esta patologia16. Este gene codifica a proteína max, envolvida na proliferação e diferenciação celular3,16. A doença só é manifesta se a mutação tiver transmissão paterna (tem imprinting materno)3,16. Pelos estudos existentes, que ainda são escassos, os casos devidos a esta mutação parecem não ter grande potencial de malignidade e produzem sobretudo noradrenalina, apesar de terem alguma capacidade de produção de adrenalina16. Existem tumores bilaterais ou múltiplos em cerca de 30% dos casos e a idade de diagnóstico é menor que os casos esporádicos sem esta mutação (idade média de 34 anos)16. PGL toracoabdominais são tão comuns como nos casos causados por mutações nos genes SDHB e SDHD, mas grande parte dos doentes portadores destes tumores têm também FEO que habitualmente é diagnosticado antes e pode ser mulficocal ou bilateral16.
Existem ainda outras mutações muito raras. Uma diz respeito ao gene KIF1B, que foi identificada em poucas famílias3. Outra refere‐se à mutação no gene PHD2, identificada apenas numa família3. Existe ainda uma outra, mais recentemente documentada em doentes com policitemia congénita, que depois vieram a desenvolver FEO ou PGL: mutação no gene HIF2A17. Estes genes não são por isso pesquisados na prática clínica3.
Vias da tumorigéneseOs estudos de expressão genética dos genes envolvidos nas síndromes associadas a FEO/PGL permitem a sua divisão em 2 categorias, tendo em conta os mecanismos que levam ao desenvolvimento tumoral11. As síndromes devidas a mutações de SDH e VHL estão associadas a angiogénese, hipoxia, reforço da matriz extracelular e redução da expressão de componentes da resposta oxidativa e do ciclo de Krebs11. Os tumores ligados à NF1 ou a mutações no gene RET mostram uma desregulação do metabolismo adrenérgico, síntese proteica e via de sinalização da cinase3,11. Tendencialmente, os FEO e PGL com mutações nos genes VHL e SDH produzem sobretudo noradrenalina, enquanto os que apresentam os genes da NF 1 e RET mutados são sobretudo adrenérgicos3.
Indicações para testes genéticosSíndrome familiar suspeita ou confirmadaHá pelo menos 2 razões para se realizar estudo genético em doentes com síndrome familiar suspeita ou confirmada. Primeiro porque as síndromes associadas a FEO/PGL acarretam risco de surgirem outros tumores e, identificando‐se os doentes portadores das mutações, pode realizar‐se uma vigilância clínica mais adequada, obtendo‐se diagnósticos mais precoces, de forma a instituir tratamento mais cedo3,5,6. Depois, porque estas síndromes genéticas se associam a formas múltiplas, recorrentes e por vezes malignas de FEO e PGL, pelo que uma vigilância apertada destes doentes permite melhorar‐lhes o prognóstico, acontecendo o mesmo com os seus familiares6.
O primeiro passo para identificar estes doentes é a realização de uma história clínica e exame físico adequados6. Deste modo, se forem identificados dados que façam suspeitar de síndromes familiares o teste genético deve ser efetuado6. Esses dados dizem respeito a história familiar positiva, a história pessoal de características relativas às várias síndromes anteriormente descritas, bem como alterações ao exame objetivo (tabela 3)6,7. As mutações são encontradas em 100% dos doentes sindrómicos e em 41‐64% dos doentes não sindrómicos, mas com história familiar positiva6. Uma vez detetada a mutação, outras neoplasias habitualmente associadas à respetiva síndrome devem ser investigadas no doente6. Deve pesquisar‐se a mutação nos familiares de primeiro grau, pois isso permite identificar indivíduos assintomáticos em risco de desenvolver a doença, para que se possa iniciar uma vigilância bioquímica e radiológica adequada, de forma a diminuir a sua mortalidade e morbilidade6.
Características clínicas suspeitas de FEO/PGL hereditário
| Características Clínicas |
| Feocromocitoma |
| Morte súbita (especialmente em idade jovem) |
| Hipertensão arterial, enfarte agudo do miocárdio, insuficiência cardíaca, acidente vascular cerebral, especialmente se em idade jovem ou durante a gravidez |
| Crise hipertensiva durante procedimentos ou anestesia |
| VHL |
| Quistos ou carcinomas renais ou pancreáticos |
| Massas testiculares em crianças |
| Surdez ou cegueira instalada em idade joven |
| Tumores do sistema nervoso central |
| MEN 2 |
| Carcinoma da tiroide ou bócio |
| Hipercalcemia e outras características associadas a hiperparatiroidismo primário (como litíase renal) |
| Ganglioneuromas da mucosa ou hábito marfanoide |
| PGL 1 e PGL 4 |
| Tumores da cabeça e pescoço ou abdominais (extra‐adrenais), torácicos e pélvicos, com sinais e sintomas sobretudo relacionados com efeito de massa (e não com excesso de catecolaminas) |
A maioria dos FEO/PGL surgem em doentes sem história familiar nem com outras características que façam pensar, desde início, numa das síndromes até agora descritas. No entanto, como já foi referido, muitas mutações surgem de novo, têm penetrância incompleta e imprinting materno, o que pode levar a uma expressão variável da doença em cada família6. Os estudos apresentam percentagens cada vez mais elevadas de doentes com mutações3,18,19, podendo mesmo ascender aos 39%20. A frequência de cada mutação é variável (VHL 3,5‐27%; RET 0,4‐5%; SDHD 0,8‐10%; SDHB 1,5‐10%6) e existem diferenças entre as várias áreas geográficas7,21. Assim, a questão do estudo genético nos doentes com FEO/PGL aparentemente esporádicos coloca‐se, de forma pertinente, havendo mesmo autores que defendam que todos os doentes o devem fazer22. No entanto, a pesquisa de mutações em todos os doentes e de todos os genes envolvidos é dispendiosa, pelo que é necessário encontrar uma forma racional de o fazer e analisar caso a caso, pesquisando os genes por ordem decrescente de prioridade3,4,23. Há situações em que o estudo genético é indiscutível: doentes com diagnóstico em idade jovem (menos de 45 anos), já que nos casos familiares a doença tende a manifestar‐se mais cedo); doença bilateral, multifocal, recorrente ou maligna; todos os PGL, especialmente os da cabeça e pescoço3,5,6,21.
A NF 1 tem alterações muito típicas, pelo que, se forem reunidos os critérios de diagnóstico (tabela 1), o teste genético não é necessário6. A pesquisa dos restantes genes tem de ser baseada nas características de cada doente, no perfil bioquímico da doença (tabela 2) e nas características histológicas do tumor6.
Doentes com FEO e PGL malignos apresentam principalmente mutações no gene SDHB, ao contrário dos doentes com mutações nos genes SDHD e SDHC, em que menos de 5% apresentam características de malignidade13,14,18. Na NF 1 identificou‐se uma percentagem de FEO maligno semelhante à da população com doença esporádica6. Os doentes com FEO ligados às síndromes VHL e MEN 2 apresentam menos risco de doença maligna que a população com doença esporádica. Deve ressalvar‐se, no entanto, que as crianças com MEN 2B tem maior risco de doença maligna em relação aos que têm MEN 2A6,24,25.
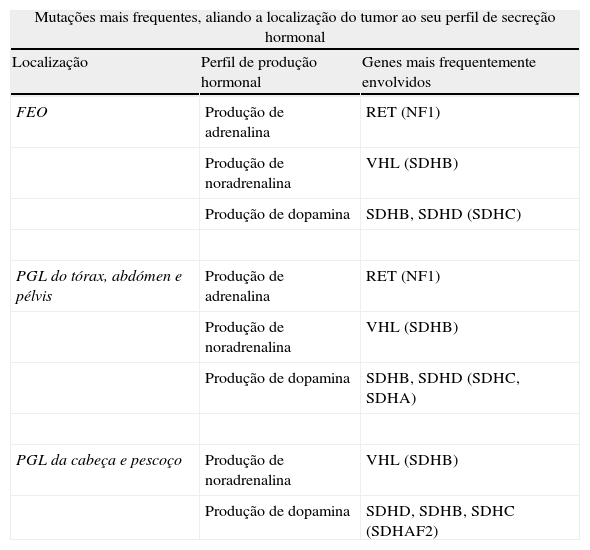
Assim, à luz de tudo o que foi descrito, parece vantajoso pesquisar as mutações nos genes SDHB, VHL e SDHD em doentes com FEO maligno ou com PGL3. Nos doentes com FEO, a realização do teste genético deve ser ditada pela idade do doente à data de diagnóstico3. Se o doente tiver menos de 45 anos devem pesquisar‐se mutações nos genes VHL, RET, SDHB e depois SDHD3. Na figura 2 está exposta uma forma de hierarquização do teste genético mais adequado em cada caso (tabela 4).

Características dos vários FEO/PGL, de acordo com a sua localização e perfil bioquímico, referindo‐se as mutações mais vezes envolvidas em cada um dos casos, de forma decrescente de probabilidade (entre parêntesis então as menos frequentes)
| Mutações mais frequentes, aliando a localização do tumor ao seu perfil de secreção hormonal | ||
| Localização | Perfil de produção hormonal | Genes mais frequentemente envolvidos |
| FEO | Produção de adrenalina | RET (NF1) |
| Produção de noradrenalina | VHL (SDHB) | |
| Produção de dopamina | SDHB, SDHD (SDHC) | |
| PGL do tórax, abdómen e pélvis | Produção de adrenalina | RET (NF1) |
| Produção de noradrenalina | VHL (SDHB) | |
| Produção de dopamina | SDHB, SDHD (SDHC, SDHA) | |
| PGL da cabeça e pescoço | Produção de noradrenalina | VHL (SDHB) |
| Produção de dopamina | SDHD, SDHB, SDHC (SDHAF2) | |
Muitos estudos estão em curso com o objetivo de encontrar formas de encontrar mais mutações e métodos mais baratos de realizar os estudos genéticos mais adequados3. Por imuno‐histoquímica já é possível distinguir os FEO/PGL com mutações ligadas aos genes do complexo SDH e outras síndromes familiares (MEN 2, VHL, NF 1) dos tumores esporádicos com elevada sensibilidade e especificidade6,26. Assim, devido aos constantes avanços da ciência, pode ser vantajoso o armazenamento do DNA do doente para futuros estudos3.
ConclusãoOs avanços recentes na genética revelaram uma nova problemática no que diz respeito aos FEO e aos PGL. Muitas mutações têm vindo a ser identificadas, fazendo aumentar a percentagem de casos familiares, sendo que grande parte deles existe nos doentes com tumores aparentemente esporádicos. Isto traz vantagens pois ao identificar a mutação pode‐se adequar a vigilância e o tratamento dos doentes, bem como a vigilância, diagnóstico precoce e tratamento atempado dos seus familiares. No entanto, fica ainda por encontrar a forma mais adequada de vigiar os indivíduos portadores da mutação, já que o facto de a terem não significa a certeza de que doença se vai manifestar.
Conflito de interesesOs autores declaram não haver conflito de interesses.
Os autores agradecem a Ana Saraiva, António Esteves e Luísa Marques pela colaboração na elaboração dos desenhos presentes neste artigo.
