








¿ Introducción
El término pancreatitis autoinmune (PAI) fue acuñado por Yoshida y colaboradores en 1995 para referirse a una forma de pancreatitis crónica de posible etiología autoinmune, caracterizada por manifestaciones clínicas, histológicas y serológicas específicas, que particularmente han mostrado respuesta al tratamiento con corticoesteroides.1 La enfermedad sistémica esclerosante asociada a IgG4, es una patología que afecta múltiples órganos, dentro de los más frecuentemente involucrados están el páncreas, glándulas salivales, lagrimales, tiroides, vía biliar, retroperitoneo y ganglios linfáticos. La afección pancreática en esta entidad se ha clasificado como PAI tipo 1.2-4
¿ Epidemiología
En un estudio japonés con 900 individuos, se estimó una prevalencia de 0.82 por cada 100 000 individuos.5,6 En Estados Unidos se encontraron hallazgos histológicos característicos de PAI en 2.4% de todas las resecciones pancreáticas,2 y hasta en 6% a 8% de las resecciones por sospecha de cáncer. Constituye de 5% a 6% de todos los casos con pancreatitis crónica.3,6,7 Es más frecuente en hombres, con un cociente hombre-mujer de 2.85. Se presenta alrededor de la quinta década de la vida, siendo 85% de los pacientes mayores de 50 años.1,6
¿ Clasificación
La PAI se clasifica histológicamente en dos tipos:
PAI Tipo 1 o pancreatitis esclerosante linfoplasmocitica (PELP), es la forma más frecuente y se presenta a mayor edad. Se caracteriza por infiltrado linfoplasmocitico periductal rico en células IgG4, fibrosis estoriforme y flebitis obliterante.4,8 La afección extra pancreática muestra hallazgos histológicos similares.9 Con mayor prevalencia de incremento de IgG4, más probabilidades de afección extra pancreática, respuesta a esteroides y recaídas.10
PAI tipo 2 o pancreatitis ductal central idiopática (PDCI), no parece estar relacionada con incremento sérico de IgG4 ni involucrar otros órganos y la respuesta a esteroides es menos clara.4 Una fracción de pacientes con PAI tipo 2 tiene enfermedad inflamatoria intestinal y reporte de esclerosis múltiple, y se ha encontrado asociación con otras enfermedades autoinmunes como síndrome de Evans y tiroiditis de Hashimoto.11
¿ Fisiopatología
La patogenia de la PAI se desconoce. Se considera que existe susceptibilidad genética asociada al haplotipo HLA-DRB1*0405-DQB1*0401 en la clase II.1
Se presume que los antígenos de HLA-DR en las células ductales y acinares pancreáticas pueden servir como antígenos reconocidos, llevando a la inflamación subsecuente. Un informe reciente describió la asociación genética de los polimorfismos del receptor 3 de Fc con la pancreatitis autoinmune.12
La mayoría de los pacientes con PAI tienen niveles elevados de IgG sérica, específicamente niveles elevados de IgG43 la cual parece desempeñar un papel principal en la patogenia.13 Otros autores proponen que IgG4 puede representar simplemente una respuesta secundaria a un disparador primario todavía no identificado del proceso inflamatorio, el mecanismo exacto se desconoce.1
¿ Cuadro clínico
La PAI se presenta con una amplia variedad de síntomas. Las manifestaciones pancreáticas suelen semejar cáncer pancreático, pancreatitis aguda, pancreatitis crónica dolorosa o insuficiencia pancreática funcional.2,4,13 El síntoma inicial suele ser ictericia de tipo obstructivo en 65% a 80%, la cual es debida a una estenosis de la vía biliar intrapancreática por inflamación pancreática de manera difusa monomórfica o por segmentos largos,2,9 cuya severidad fluctúa. La intolerancia a la glucosa y diabetes mellitus tipo 2 se han encontrado en 45% a 76% de los casos, la cual suele mejorar después del tratamiento con esteroide.4,6,7
Frecuentemente se detecta cierta disfunción exocrina pancreática de leve a moderada.6 Otros síntomas pueden incluir: dolor abdominal leve inespecífico en 35%, disminución de peso en 35% y anorexia.6,7
Manifestaciones extra pancreáticas: Pueden presentarse años antes, al mismo tiempo o después de la afección pancreática. Los órganos comúnmente afectados incluyen: vía biliar, glándulas salivales, retroperitoneo, ganglios linfáticos, parénquima renal, orbita, pulmón y próstata.4
La vía biliar es la más afectada presentándose en 70% a 100% de los casos; se conoce como colangitis esclerosante asociada a IgG4.10 La afección hepática es frecuente, la cual puede deberse tanto a lesiones extra hepáticas obstructivas o a daño inflamatorio hepático. Se han descrito cinco patrones histológicos: inflamación portal con o sin hepatitis de interfase, obstrucciones largas biliares, esclerosis portal, hepatitis lobular y colestasis canalicular.9,10 La afección renal comprende insuficiencia renal leve o lesiones múltiples de baja atenuación. La vesícula suele presentar colecistitis difusa alitiásica. En el tubo digestivo suele haber células plasmáticas IgG4 presentes en la mucosa. Se ha encontrado PAI hasta en 17% en pacientes con colitis ulcerativa crónica inespecífica y en menor porcentaje enfermedad de Cronh.1
La afección en glándulas salivales y lagrimales incluye el tumor de Küttner o sialoadenitis esclerosante crónica que se presenta como tumefacción asimétrica de las glándulas submandibulares.10 Ocasionalmente se observa síndrome de Sjögren, el cual muestra niveles de IgG4 menores. La enfermedad de Mikulicz se caracteriza por secreción idiopática bilateral y dolor de glándulas lagrimal, parótida y submandibular. La afección a retroperitoneo y mesenterio incluye fibrosis retroperitoneal. Se ha señalado que hasta 25% de los pacientes con PAI padecen hipotiroidismo clínicamente significativo.4 El compromiso pulmonar puede presentarse como neumonía intersticial, pseudotumor inflamatorio, nódulos difusos, infiltrados y adenopatías; la linfadenopatía concomitante es común.1,4
¿Hallazgos de laboratorio
La mayoría de los pacientes con PAI, muestran niveles normales o ligeramente elevados de amilasa y lipasa. En las pruebas de función hepática se puede encontrar un perfil colestásico.7 También se ha encontrado hipergammaglobulinemia mayor a 2 g/dL en 53% a 71%;3 existe eosinofilia periférica con más de 600 células/mm en 11%,1 e incremento de IgE en 34%.5
¿Estudios de imagen
Ultrasonido (USG) abdominal: La imagen pancreática en un USG abdominal es subóptima. En la PAI el páncreas se observa con hipoecogenicidad difusa y engrosamiento del conducto biliar.4
Tomografía computarizada (TC): El parénquima pancreático está difusamente agrandado, en forma de salchicha con atenuación homogénea (Figura 1). Con la aplicación del medio de contraste se observa realce tardío, prolongado y un borde tipo cápsula de baja densidad rodeando la glándula (halo de hipoatenuación).1-4 Aún si no se observa el halo de hipoatenuación, el agrandamiento con forma de salchicha con realce retardado es altamente sugestivo de PAI. Además el hallazgo de estrechamiento difuso del conducto pancreático es altamente diagnóstico de PAI.1,4,13 Cuando la participación es focal más que difusa, ésta se localiza con más frecuencia en la cabeza del páncreas observándose un agrandamiento focal y de forma menos común una masa de baja atenuación o isoatenuación.1,4 Tanto en la forma difusa como en la focal no se encuentra dilatación del conducto pancreático ni atrofia del páncreas.4 También es común encontrar crecimiento de ganglios linfáticos regionales.6,13
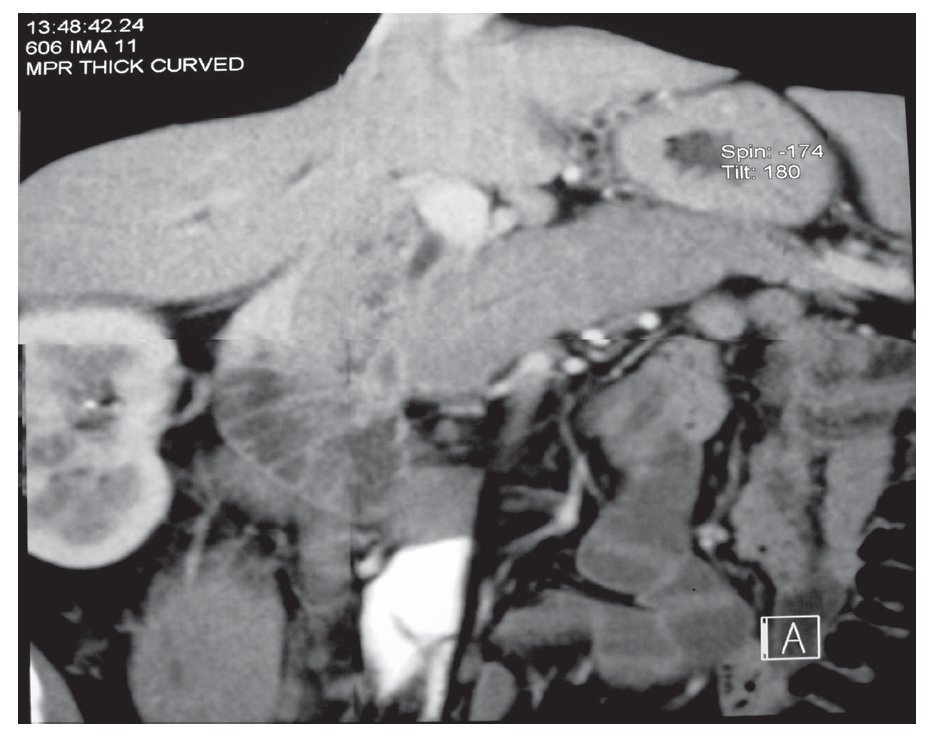
¿ Figura 1. TC de abdomen de un paciente con PAI en el que se observa la imagen de "páncreas en salchicha.
Los hallazgos extrapancreáticos son: dilatación focal de los conductos biliares intrahepáticos y extrapancréaticos, adenopatía intraabdominal voluminosa, alteraciones del parénquima renal y fibrosis retroperitoneal.4
Resonancia magnética (RM)/Colangiopanceratoresonancia (CPRM): Se observa un páncreas agrandado con disminución en la intensidad de señal e incremento en la intensidad de la señal en las imágenes ponderadas en T2 y ocasionalmente un borde paripancreático hipointenso (Figura 2).2-4,13 Se puede encontrar una atenuación difusa o una estenosis pequeña del conducto pancreático principal.1
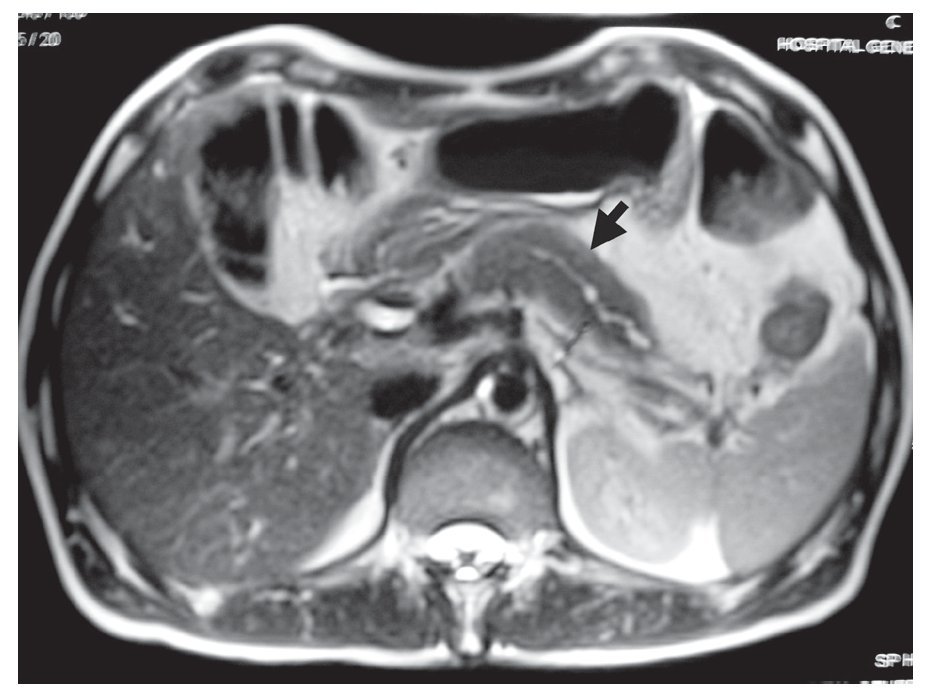
¿ Figura 2. Resonancia magnética de un paciente con pancreatitis autoinmune en la que se observa aumento del tamaño del páncreas.
Colangiopancreatografía retrograda endoscópica (CPRE): El hallazgo característico es una atenuación focal, segmentaria o difusa del conducto pancreático principal y la desaparición de ramas en ángulo recto. El conducto pancreático principal adyacente al área de estenosis esta mínimamente dilatado. El conducto biliar también puede estar afectado, observándose un estrechamiento de la porción intrapancreática del conducto biliar común, estrechamiento irregular del conducto biliar extrahepático y con menos frecuencia dilatación de los conductos biliares intrahepáticos. La afectación del conducto biliar intrapancreático semeja pancreatitis crónica o cáncer pancreático, y la participación del conducto biliar proximal e intrapancreático semeja un colangiocarcinoma y colangitis esclerosante primaria (CEP). La imagen de la PAI focal es muy similar a la apariencia del cáncer pancreático.3,4,7 Nakazawa y colaboradores señalaron que la colangiografía es un método eficaz para diferenciar PAI de CEP.14
Ultrasonido Endoscópico (USE): Permite visualizar el páncreas y los conductos pancreático y biliar, y proporciona una oportunidad para obtener aspirado por aguja fina (AAF) y tomar biopsias con aguja gruesa para el diagnóstico histológico. El valor predictivo negativo de AAF con USE para cáncer pancreático es de aproximadamente 75% y, las biopsias con aguja gruesa son diagnósticas de PAI en aproximadamente 75% de los pacientes.1-4
¿ Serología
Aunque los niveles de IgG4 séricos pueden ser normales aun en presencia de hallazgos histológicos clásicos de PAI, a menudo los pacientes con PAI tienen un incremento en el nivel de la IgG4 sérica.2,3,13 Hamano y colaboradores fueron los primeros en establecer el valor diagnóstico de la IgG4 sérica como marcador para PAI, ellos reportaron que los niveles séricos de IgG4 son altamente sensibles (95%) y específicos (97%).15 Ghazale y colaboradores encontraron que un valor de IgG4 mayor de 140 mg/dL tiene una sensibilidad de 76%, especificidad de 93% y valor predictivo positivo de 36% para el diagnóstico de PAI y cuando el punto de corte fue dos veces el valor normal de IgG4 (más de 280 mg/dL) los valores fueron 53%, 99% y 75% respectivamente.16 En cuanto a sensibilidad, Deheragoda y colaboradores señalaron un valor de 45.5%,17 Raina y colaboradores de 44%18 y Tae Jun Song de 52.4%.12 La especificidad de la IgG sérica total se informa de 95%, lo cual no fue significativamente diferente de la IgG4 sérica (90%) en la diferenciación de la PAI del cáncer pancreático.16 Ohara y colaboradores señalaron que los niveles séricos de IgG4 fueron significativamente más altos en pacientes con PAI con lesiones sistémicas extrapancreáticas que aquellos que no las presentaban.19
En un estudio se encontró que la medición combinada de la IgG total y de la IgG4 sérica puede incrementar la sensibilidad diagnóstica sin sacrificar la especificidad, comparada con la IgG4 sola. La sensibilidad de la IgG4 mayor de 135 mg/dL fue de 52.5% significativamente mayor que IgG total superior de 1800 mg/dL. La sensibilidad de la medición combinada de IgG total e IgG4 fue de 68.3%, significativamente mayor que la IgG sola. La especificidad de la medición combinada de la IgG total e IgG4 fue de 95.5% y no fue significativamente diferente de la IgG4 sola.12
Otras anormalidades serológicas encontradas son hipergammaglobulinemia más de 2 g/dL y niveles séricos de IgG más de 1800 mg/dL en 53% a 76% y 53% a 71% de los pacientes con PAI, respectivamente. Los autoanticuerpos que se han detectado en pacientes con PAI incluyen anticuerpos antinucleares 44% y factor reumatoide (FR) 16%, anticuerpos antilactoferrina, anticuerpos anti anhidrasa carbónica II y anticuerpos antimúsculo liso.2,3 Tae Jun Song y colaboradores informaron una sensibilidad de 24.4% y 20.3% para ANA superior a 1:80 y FR respectivamente.12 Asada y colaboradores recientemente propusieron que el anticuerpo anti-inhibidor de secreción del tripsinógeno pancreático de la subclase IgG1 puede ser un marcador diagnóstico altamente específico para diferenciar PAI de otras enfermedades pancreáticas, ellos detectaron este anticuerpo en 43% de los pacientes con PAI y en ninguno de los controles sanos.20
¿ Histopatología
En su aspecto macroscópico, el proceso fibroinflamatorio dentro del páncreas puede semejar un adenocacinoma ductal, localizado comúnmente en la cabeza del páncreas. Ocasionalmente la fibrosis está en el cuerpo o cola del páncreas y también puede encontrarse la participación difusa de la glándula. La arquitectura lobular normal se pierde y algunas veces se observa fibrosis infiltrando el tejido blando peripancreático. Cuando la PAI involucra la cabeza del páncreas, la participación del conducto pancreático principal resulta en estenosis proximal, obstrucción y dilatación distal; en más de 90% de los casos el conducto biliar común está involucrado y aparece engrosado, estenótico y con dilatación proximal.2,3
En la PELP hay tres hallazgos histológicos esenciales: 1) infiltrado linfoplasmocítico del espacio periductal con células plasmáticas (frecuentemente IgG4 positivas) y linfocitos T, 2) fibrosis estoriforme alrededor de los conductos interlobulares grandes y medianos y 3) flebitis obliterativa en donde el infiltrado rodea y oblitera las venas pancreáticas.7 La inmunotinción revela abundantes células IgG4 positivas mayor de 10 células por campo de alto poder (Figura 3). Se evidencian macrófagos y eosinófilos dispersos.2,3,8,13 El hallazgo de venulitis obliterativa junto con la inflamación linfoplasmocitica periductal, se considera diagnóstica de PAI.3
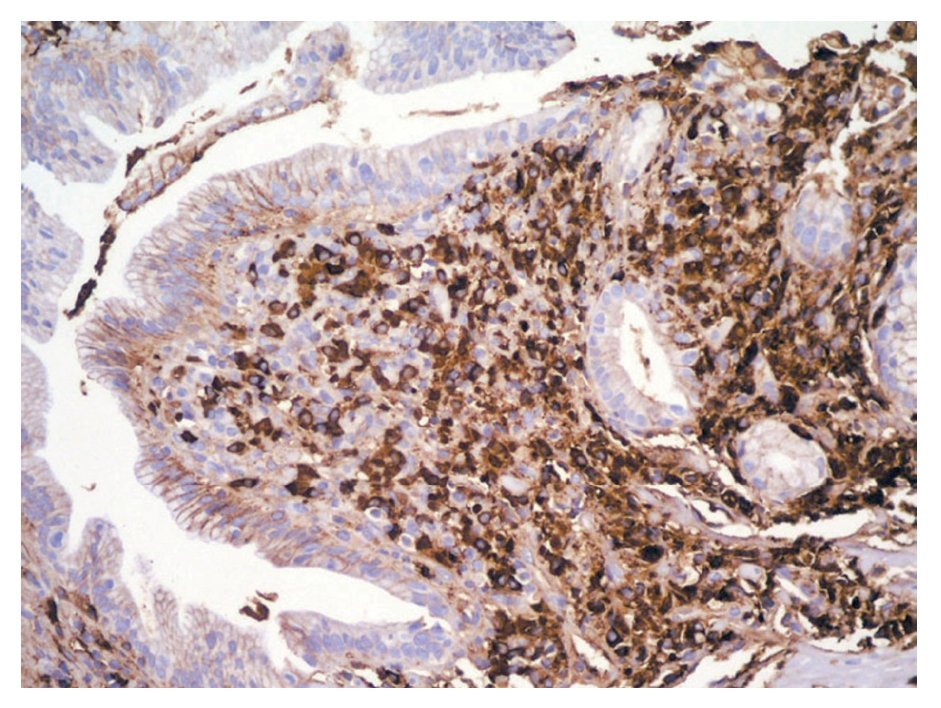
¿ Figura 3. Inmunohistoquimica en biopsia de ámpula de Vater en la que se observan células plasmáticas positivas para IgG4.
Zhang y colaboradores dividen el infiltrado IgG4 positivo en leve (menos de 10/campo de alto poder [hpf]), moderado (11 - 30/hpf) e importante (más de 30/hpf). Ellos encontraron que los pacientes con PAI frecuentemente tienen un infiltrado por células plasmáticas IgG4 positivas moderado o importante en el tejido pancreático. Una característica diagnostica de PAI es la presencia de una infiltrado linfoplasmocítico con más de 10 células plasmáticas IgG4 positivas/ hpf en el páncreas.21
En la pancreatitis ductocéntrica idiopática (PDI) hay infiltrado linfoplasmocítico periductal. La inflamación difusa, la fibrosis estoriforme y la flebitis obliterativa son menos prominentes que en PELP.
El hallazgo más distintivo de PDI es la presencia de lesión granulocítica en los conductos de mediano y pequeño tamaño y en los acinos. El otro hallazgo distintivo es la presencia escasa (menos de 10 cels/hpf) o la completa ausencia de células plasmáticas positivas para IgG4 en la inmunotinción.8
¿ Criterios diagnósticos
El estándar de oro para el diagnóstico de PAI es la histología de PELP con inmunohistiquímica positiva para IgG 4 del tejido pancreático (más de 10 cels/hpf). La presencia de todo el espectro de LPSP en la histología requiere biopsia con aguja gruesa porque la aspiración con aguja fina usualmente no es suficiente para hacer el diagnóstico. Dado que el páncreas no es fácilmente accesible a la biopsia con agua gruesa, los criterios tratan de proponer criterios sustitutos para limitar la biopsia solo a los casos más difíciles.1,2,4
Las primeras guías reales para el diagnóstico de PAI fueron las de la Sociedad Japonesa de Páncreas (SJP) en 2002 posteriormente modificadas en 2006. El principal objetivo de esos criterios fue asegurar que se excluyera el diagnóstico de cáncer de páncreas.22 En 2006, Chiari y colaboradores propusieron los criterios HISORt de la clínica Mayo.23 En 2008 la SJP y la sociedad coreana de enfermedades pancreatobiliares propusieron los criterios del consenso Asiático en donde se propuso eliminar la gammaglobulina de los criterios serológicos.24
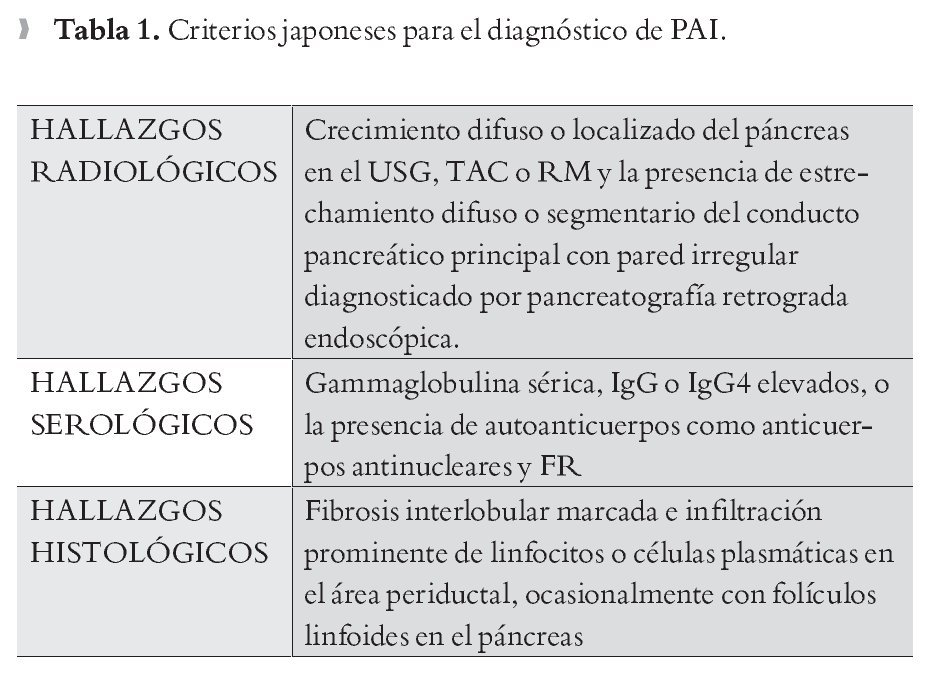
Criterios Japoneses: Para hacer el diagnóstico de PAI basado en las guías Japonesas se requiere que los hallazgos radiológicos sean consistentes con PAI y uno de los criterios serológicos o histológicos (Tabla 1).2,22
Criterios HISORt HISORt es una nemotecnia para Histología, Radiología, Serología, Otros órganos involucrados y Respuesta al tratamiento. Los pacientes son diagnosticados como PAI si se encuentran en uno de tres grupos (Tabla 2).23
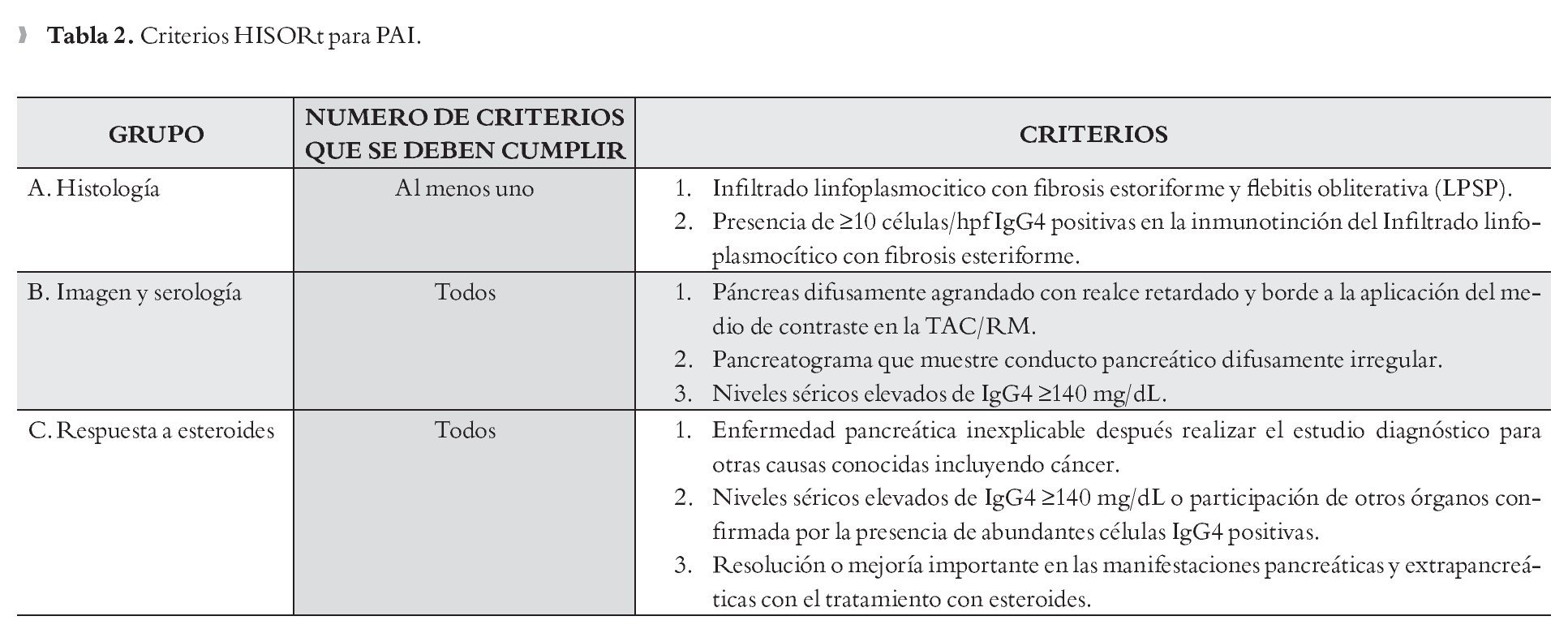
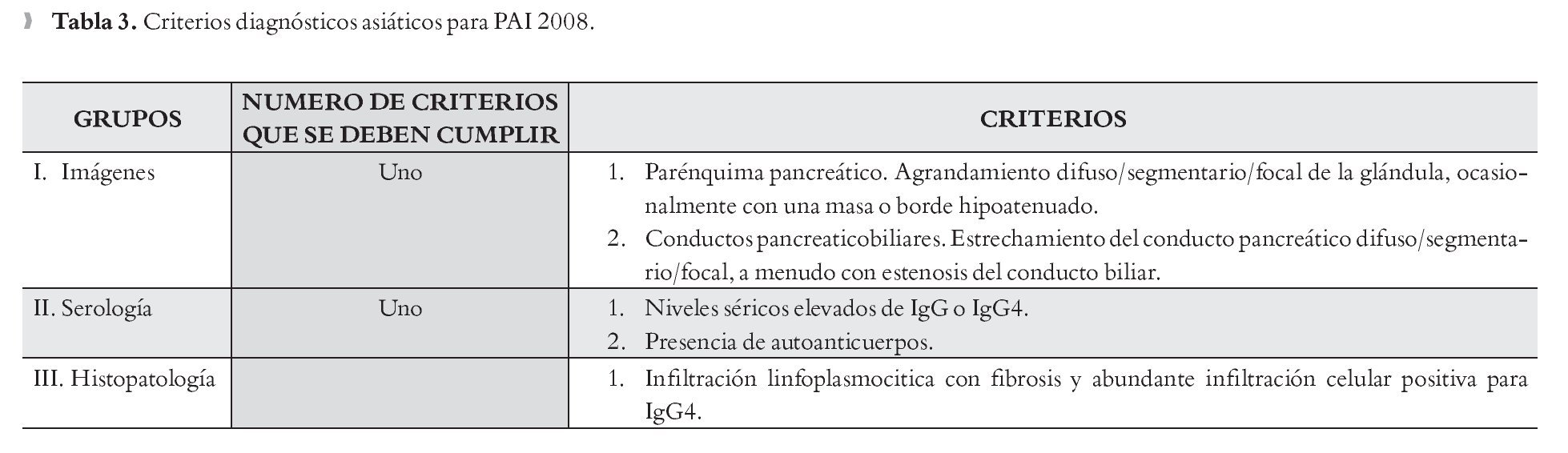
Criterios Diagnósticos Asiáticos para PAI 2008: Se puede hacer el diagnóstico de PAI si se cumple el criterio I además de uno de los criterios II o por el criterio III solo (Tabla 3).24
¿ Tratamiento
El tratamiento de primera elección es la terapia con esteroides, mostrado mejoría de los síntomas, regresión del proceso inflamatorio y resolución de anormalidades radiográficas y laboratorio.3,7,25
Las dosis de esteroides recomendadas no han sido estandarizadas.3,7 Algunos autores consideran la terapia inicial con 30 a 40 mg/día de prednisona por cuatro semanas, seguidas de una disminución gradual 5 a 10 mg/semana de cuatro a seis semanas con una dosis de mantenimiento 5 a 10 mg/día y continuar hasta resolverse completamente las anormalidades clínicas, de laboratorio o radiográficas.3,7,23,25
Aunque la mayoría de los pacientes tienen una respuesta rápida en pocas semanas, un grupo pequeño podría requerir de terapia de mantenimiento con prednisona a una dosis de 5 mg a 10 mg por día.1,3 La terapia de mantenimiento con bajas dosis de esteroides es usada para prevenir las recaídas y mantener la remisión. La duración de la etapa de mantenimiento no se ha establecido.25
La respuesta a esteroides se evalúa de acuerdo a los síntomas antes y después al uso de esteroides, medición de enzimas hepáticas, niveles de IgG/IgG4 y la apariencia radiológica.1,3,25
El tratamiento con esteroides en pacientes asintomáticos pero con anormalidades radiológicas no está claro aún, se ha visto mayor número de recaídas en pacientes asintomáticos no tratados que aquellos asintomáticos que recibieron corticoesteroide, esto justificaría el iniciar tratamiento en todos los pacientes.7
Aun no se ha definido si los pacientes con estenosis se beneficiarían con esteroides o con la colocación de prótesis y posteriormente dar esteroides. Estas prótesis podrían ser removidas dos meses después de iniciar el tratamiento.7,25 Los pacientes que presentan ictericia por estenosis biliar en los cuales no es posible la colocación de protesis, pueden ser tratados con un bolo de corticoesteroides.7
El seguimiento debe ser periódico para la identificación de una recaída temprana, este debe incluir manifestaciones clínicas (ictericia, dolor abdominal o molestia), examenes de laboratorio, niveles de IgG4 e estudios de imagen.25
Hasta el momento no existe consenso acerca de la definición de remisión y recaída. La recaída puede ser sintomática, radiológica, serológica e histológica.25,26
La recaída después de la remisión ocurre en una tercera parte de los pacientes. La manifestación clínica más común es la estenosis biliar.25 De los pacientes 30% a 40% con recaída clínica o radiológica requiere retratamiento con un segundo curso prolongado de esteroides.2,7 En la recaída con un segundo curso de esteroides es necesario utilizar otros agentes como azatioprina o 6-mercapturina,7 los cuales han mostrado remisión a seis meses de seguimiento.4
La identificación de predictores clínicos o genéticos de recaída es crucial para el pronóstico y evolución clínica de la enfermedad y podrían ayudar a determinar si un paciente podría beneficiarse realmente con terapia de mantenimiento, ajustar dosis, incremento de la dosis o administración por mayor tiempo.25 Varios autores entre ellos Hinaro, Park, Kubota y Chazale han escrito acerca de predictores clínicos en recaídas, la mayoría de ellos le dan peso a la edad, género, ictericia obstructiva, niveles de IgG4, así como cambios patológicos.27-30
Se han reportados como predictores genéticos de recurrencia la sustitución de ácido aspártico a acido no aspártico (alanina, valina y serina) en DQB1 57, presencia de HLA y polimorfismo del antígeno cuatro de los linfocitos T citotóxicos (CTLA4).25,26 Predictores séricos como el factor reumatoide monoclonal (MRF), con un valor de corte de 10 μg/dL sirve como predictor de recurrencia con sensibilidad 61% y especificidad de 70%. La probabilidad de recurrencia es de 60% con valores por arriba de 10 μg/dL y de 30% valores menores de 10 μg/dL.26
El pronóstico a largo plazo se desconoce.2 Hirano y colaboradores realizaron un estudio retrospectivo para valorar el pronóstico de los pacientes con pacientes con PAI que reciben tratamiento con corticoesteroides comparado con aquellos que no reciben tratamiento. De los 19 pacientes que recibieron esteroides 32% tuvieron una evolución desfavorable presentando neumonia intesticial o ictericia obstructiva. De los 19 pacientes que no recibieron tratamiento con corticoesteroides, 70% tuvieron eventos desfavorables que incluian ictericia obstructiva por estenosis del conducto biliar distal, crecimiento de pseudoquiste, cambios esclerogénicos del conducto biliar extrapancreático, hidronefrosis por fibrosis retroperitoneal y nefritis intersticial. Con estos resultados los autores concluyen que el tratamiento con corticoesteroides puede reducir los eventos desfavorables relacionados con PAI.27 Aunque el estado infalamatorio crónico puede incrementar el riesgo a cáncer pancreático, solo se han reportado dos casose cáncer en pacientes con PAI.4,25
Correspondencia: Dra. Nashiely Gil Rojas.
Dr. Balmis 148. Col. Doctores. Delegación Cuauhtémoc. México, D. F. 06726.
Teléfono: 2789 2000, ext. 1044.
Correo electrónico: nagirs240681@yahoo.com.mx.