



La miositis osificante es un trastorno benigno caracterizado por la formación de hueso heterotópico en el músculo esquelético o en los tejidos blandos. Es extremadamente rara en niños, <1% de los casos ocurren en menores de 10años.
Presentamos el caso de un niño de 17días que, tras 10días de ingreso en una Unidad de Cuidados Intermedios, fue derivado a nuestro hospital por una displasia del desarrollo de la cadera. En el examen clínico presentaba tumefacción y aumento del tamaño del muslo izquierdo respecto al contralateral, por lo que ingresó para estudio. Se describen los hallazgos en las imágenes de radiografías simples, tomografía computarizada, resonancia magnética y gammagrafía ósea, así como el tratamiento realizado. Hasta donde sabemos, es el caso más joven reportado en la literatura.
Myositis ossificans is a benign disorder characterized by the formation of heterotopic bone in skeletal muscle or soft tissues. It is extremely rare in children, <1% of cases occur in children under 10 years.
We present a 17-day-old boy that, after 10 days of Intermediate Care Unit stay, was referred to our hospital for a developmental dysplasia of the hip. On clinical examination, he had swelling on the left thigh and increase in size compared to the contralateral one, therefore was admitted for studying. Imaging findings including plain radiographs, CT, MRI and bone scintigraphy, as well as treatment performed, are described. To the best of our knowledge, is the youngest case reported in the literature.
La miositis osificante (MO) es una enfermedad benigna que consiste en la formación de hueso heterotópico en los músculos o tejidos blandos1. Esta entidad bien definida se puede dividir en tres tipos: miositis osificante progresiva (fibrodisplasia osificante progresiva [FOP]), miositis osificante circunscrita o traumática (MOT) y miositis osificante no traumática o pseudomaligna (MOP)2. La etiología de esta última forma no se comprende bien a día de hoy. Cualquier parte del cuerpo se puede afectar, pero la zona anterior del muslo es el sitio de aparición más frecuente3. En las primeras etapas, las lesiones de MO pueden presentarse de forma similar a un rabdomiosarcoma, osteosarcoma, o linfoma1. Habitualmente afecta a adolescentes y adultos jóvenes, con un ligero predominio del sexo masculino2,3.
Se presenta el caso de un lactante de 17días de edad con MO, tras un periodo corto de inmovilización. Cuatro aspectos hicieron que el diagnóstico de MO fuera muy desafiante en este niño. En primer lugar, la MO es extremadamente rara en niños menores de 10años, con pocos casos reportados en la literatura y, hasta hoy, ninguno en recién nacidos. En segundo lugar, la ausencia de un traumatismo conocido no levantó sospechas hacia este diagnóstico. Sin embargo, aunque se encuentra un traumatismo mayor o menor recurrente en el 40-60% de los casos de MO, en muchos casos no hay trauma presente. En tercer lugar, la presentación clínica acompañada de otros trastornos esqueléticos y un fenotipo peculiar fueron signos y síntomas clínicos engañosos que podían situarlo en un contexto sindrómico. Por último, las imágenes obtenidas en los diferentes estudios mostraban características comúnmente encontradas en las infecciones y tumores malignos musculoesqueléticos. Cuando la MO presenta una historia característica y un patrón zonal claro en las imágenes, el diagnóstico es relativamente sencillo. Sin embargo, no es raro que la aparición de MO sugiera otras consideraciones, lo que dificulta el diagnóstico.
Caso clínicoVarón recién nacido por parto cefálico eutócico a las 41semanas, resultado del tercer embarazo de una pareja no consanguínea, con un hijo clínicamente sano y un mortinato. Embarazo controlado sin incidencias. Presentó peso adecuado para la edad gestacional, con puntuación de Apgar de 9 al minuto, y de 10 a los 5minutos. En el examen clínico destacaban características craneofaciales dismórficas, incluyendo hipertelorismo, micrognatia y orejas de implantación baja; criptorquidia bilateral, leve hipotonía axial, flexoabducción de caderas de 40° con signo de Ortolani positivo bilateral y pies en mecedora.
Se solicitó una ecografía de ambas caderas, que mostró una subluxación de la cadera derecha y una luxación de la cadera izquierda con cobertura ósea menor del 33%. Se inició tratamiento con arnés de Pavlik. Las pruebas serológicas para descartar infecciones congénitas (TORCH) y las pruebas metabólicas fueron normales. Se realizó estudio cromosómico sin hallar anomalías en el total de las metafases analizadas. Fórmula cromosómica 46XY coherente con el fenotipo masculino. Ecografías craneal, cardiaca y abdominal, sin demostrar hallazgos patológicos. Las exploraciones de oftalmología, cardiología y otorrinolaringología fueron normales.
A las 48h de vida ingresó en la unidad de cuidados intermedios (UCIN) por fiebre, ictericia idiopática, pérdida ponderal del 10% e hipernatremia sin otros signos de deshidratación. Precisó sueroterapia durante 48h con normalización de los datos analíticos, y necesitó soporte ventilatorio con oxígeno para mantener saturaciones normales. En un control analítico se apreció aumento de reactantes de fase aguda, por lo que se trató con antibioterapia intravenosa empírica hasta la llegada de hemocultivos negativos. Tras 10días de estancia en la UCIN, su estado respiratorio mejoró progresivamente desapareciendo las necesidades de oxígeno y la fiebre, y se normalizaron los parámetros analíticos de infección, por lo que se trasladó a la planta de pediatría.
A los 17días de vida se derivó a nuestro centro para valoración de la displasia del desarrollo de cadera y de los pies planos valgos convexos. En la exploración física de la cadera izquierda destacó una tumefacción de la raíz del muslo con aumento de tamaño respecto al contralateral, por lo que ingresó para completar estudios.
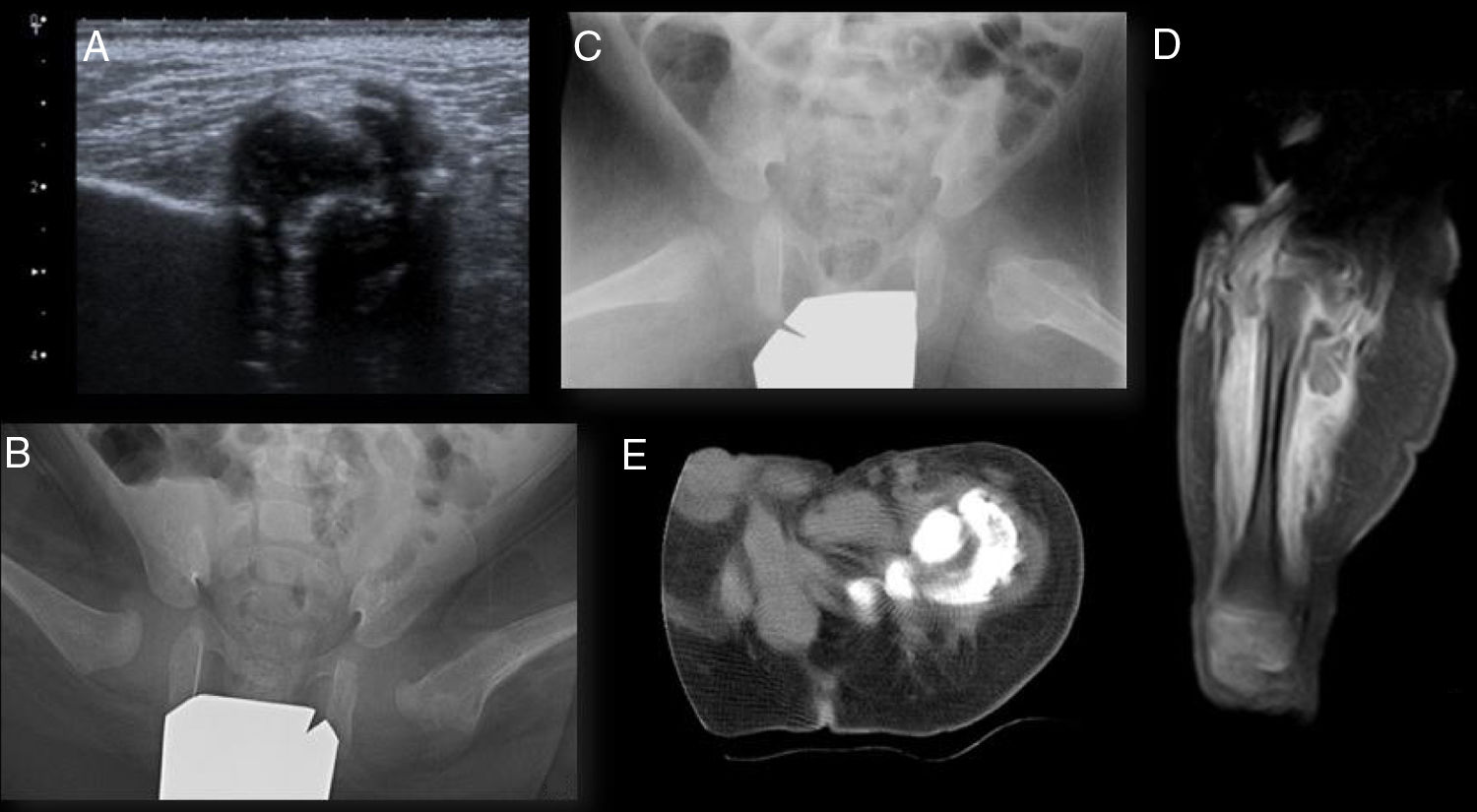
La ecografía de caderas reveló una gran calcificación en el seno de la musculatura glútea izquierda (fig. 1a). La radiografía de pelvis anteroposterior mostró una luxación de la cadera izquierda y subluxación de la cadera derecha, con una imagen de características densas en la zona anteromedial del fémur proximal izquierdo; además, en la radiografía axial de ambas caderas se observó una alteración en la cortical del fémur proximal izquierdo (figs. 1b y c). Las radiografías de los pies objetivaron un astrágalo vertical bilateral.
 Ecografía de cadera izquierda. Imagen hiperecogénica con sombra acústica posterior correspondiente a gran calcificación en el seno de la musculatura glútea izquierda. (b) y (c) Proyección anteroposterior y lateral de la rana de ambas caderas. Aumento de densidad en los tejidos blandos adyacentes a la corteza medial y lateral de la cabeza y cuello femoral izquierdo, con insuflación de la cortical en el fémur proximal. (d) Resonancia magnética cadera izquierda. Reacción perióstica continua en el fémur con edema circundante y calcificación intramuscular en el espesor de los músculos de la raíz del muslo. (e) TC cadera izquierda. Gran calcificación en el espesor de los músculos de la raíz del muslo, lateral, posterior, y medial al fémur, sin contactar con él. Una zona lúcida entre la lesión y el hueso subyacente, la presencia de una cortical intacta, la ubicación adyacente al eje de un hueso y la calcificación más densa en la periferia de la lesión son hallazgos radiográficos valiosos en el diagnóstico diferencial de malignidad ósea.")
Pruebas de imagen. (a) Ecografía de cadera izquierda. Imagen hiperecogénica con sombra acústica posterior correspondiente a gran calcificación en el seno de la musculatura glútea izquierda. (b) y (c) Proyección anteroposterior y lateral de la rana de ambas caderas. Aumento de densidad en los tejidos blandos adyacentes a la corteza medial y lateral de la cabeza y cuello femoral izquierdo, con insuflación de la cortical en el fémur proximal. (d) Resonancia magnética cadera izquierda. Reacción perióstica continua en el fémur con edema circundante y calcificación intramuscular en el espesor de los músculos de la raíz del muslo. (e) TC cadera izquierda. Gran calcificación en el espesor de los músculos de la raíz del muslo, lateral, posterior, y medial al fémur, sin contactar con él. Una zona lúcida entre la lesión y el hueso subyacente, la presencia de una cortical intacta, la ubicación adyacente al eje de un hueso y la calcificación más densa en la periferia de la lesión son hallazgos radiográficos valiosos en el diagnóstico diferencial de malignidad ósea.
Las imágenes de la resonancia magnética (RM) demostraron una infiltración de la musculatura de la raíz del muslo izquierdo asociando una calcificación intramuscular prominente, y una reacción perióstica continua en el fémur (fig. 1d) con edema circundante. La gammagrafía ósea con 99Tc-difosfonato mostró una intensa actividad osteoblástica en la región del trocánter mayor del fémur proximal izquierdo, sin apreciar otros focos periféricos. Las imágenes de reconstrucción axial y coronal de la tomografía computarizada (TC) de pelvis revelaron un aumento de volumen de toda la musculatura del muslo, especialmente cuádriceps, semitendinoso y semimembranoso, con una gran calcificación en el espesor de los músculos de la raíz del mismo, lateral, posterior, y medial al fémur, sin contactar con él, y una reacción perióstica continua, en la región lateral y posterior de la mitad superior de la diáfisis femoral (fig. 1e). Se realizó una biopsia percutánea de tejido muscular y óseo bajo control de escopia, que tuvo que repetirse y realizarse de forma abierta por ser no concluyente, con diagnóstico anatomopatológico definitivo de MO.
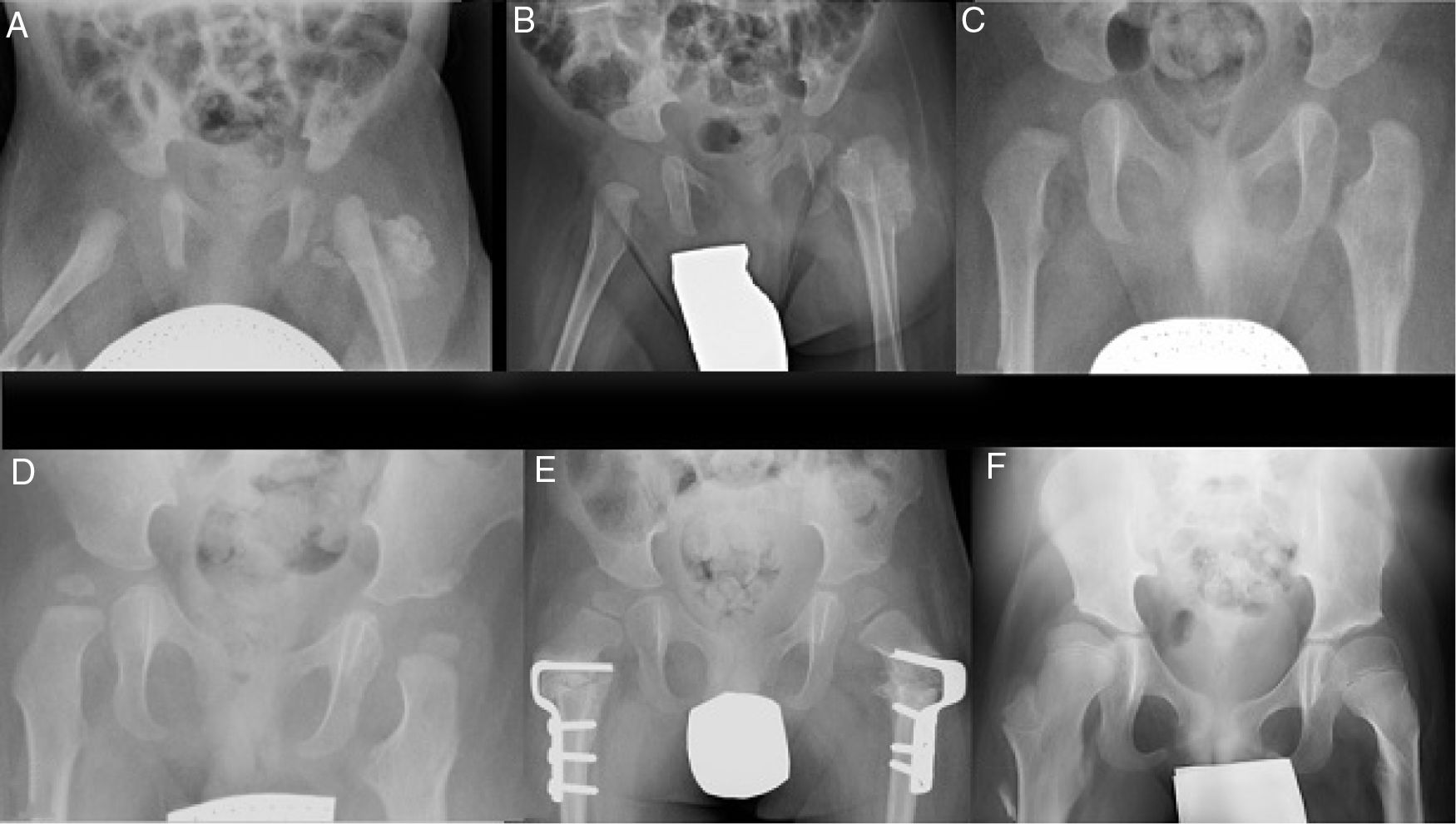
ResultadosSe decidió tratamiento conservador con estrecho seguimiento mediante radiografías convencionales seriadas (fig. 2), las cuales mostraron una alteración morfológica tanto en la metáfisis como en la cortical lateral del fémur proximal izquierdo (fig. 2c), que más tarde remodeló, junto con una disminución progresiva del tamaño de las masas osificadas hasta su completa reabsorción a los 18 meses (fig. 2d).
 Evolución de la calcificación en el espesor de la musculatura glútea y cambios morfológicos a nivel de fémur proximal al mes, 2 y 9meses, respectivamente. (d) Remodelado completo del fémur proximal a los 18meses. (e) 3años: osteotomía varizante y desrotatoria femoral proximal bilateral. (f) 8años: caderas reducidas sin evidencia de recurrencia de la lesión.")
Radiografía seriadas durante el seguimiento. (a-c) Evolución de la calcificación en el espesor de la musculatura glútea y cambios morfológicos a nivel de fémur proximal al mes, 2 y 9meses, respectivamente. (d) Remodelado completo del fémur proximal a los 18meses. (e) 3años: osteotomía varizante y desrotatoria femoral proximal bilateral. (f) 8años: caderas reducidas sin evidencia de recurrencia de la lesión.
Por su displasia del desarrollo de cadera, el paciente recibió un tratamiento ineficaz con arnés de Pavlik, consiguiendo estabilizar la cadera izquierda mediante una reducción cerrada. Los pies en mecedora se trataron con yesos seriados que no consiguieron reducir la deformidad, por lo que se realizó cirugía correctora de ambos pies al año de vida, y posterior tratamiento con ortesis de tobillo. Realizó rehabilitación y terapia de psicomotricidad y fue valorado por Neurología a los 2años, pues aún no se mantenía de pie solo, si bien el paciente había llevado yesos tras las distintas cirugías y sí intentaba ponerse en bipedestación y hacía intentos de gatear. Con sospecha de retraso motor, se derivó a un Centro de Atención Infantil Temprana. A los 2años y medio se objetivó radiológicamente un valgo del extremo proximal de ambos fémures, asociado a una subluxación de ambas caderas, por lo que se realizó una osteotomía varizante y desrotatoria femoral proximal bilateral (fig. 2e). A los 5años comenzó a deambular con andador, con 6años inició marcha con bastones precisando silla de ruedas para trayectos largos por cansancio. Actualmente el niño tiene 8años, camina ágil con muletas, presentando apoyo plantígrado de ambos pies, las caderas están reducidas, y no hay evidencia de recurrencia de la lesión osificada a nivel del fémur (fig. 2f).
DiscusiónLa MO es una entidad poco frecuente que puede aparecer a cualquier edad, pero rara vez ocurre en bebés y pacientes mayores3. El paciente más joven reportado en la literatura fue el caso de una niña de 5meses, y el más longevo, una mujer de 81años3. Nuestro paciente destaca por su corta edad, con el diagnóstico a los 17días de vida.
Las localizaciones más frecuentes de aparición de la lesión incluyen el muslo, el brazo, el hombro y la mano4,5. Es poco frecuente encontrarlo en la región paravertebral y el cuello, y en la mayoría de los casos la afectación es unilateral1,4.
La patogénesis en la MOT está bien definida, pero aún no está clara en la MOP, cuando la masa aparece en ausencia de traumatismo o evidencia de enfermedad sistémica3. En la MOT se presume que un traumatismo inicial en una región provoca una reacción fibroblástica, y posteriormente existe, en cierto grado, una metaplasia ósea y cartilaginosa2. En la mayoría de los casos no traumáticos se han implicado, como posibles factores causales, microtraumatismos repetidos, procesos isquémicos e inflamatorios3. Algunos autores argumentan que, particularmente en niños pequeños, lo más probable es que haya existido algún tipo de trauma no objetivado1,6. Estas argumentaciones están también respaldadas por el hecho de que no existen diferencias, en cuanto a la presentación clínica, imágenes y cambios histopatológicos, entre la MOT y la MOP. Por lo tanto, ambas formas se incluyen en la categoría general de MO6. Varios estudios han demostrado que la MO tiene una gran relación con la expresión de la proteína morfogenética ósea (BMP), más abundante en el plasma del niño que en el del adulto7. Así, se ha informado que la maduración y consiguiente osificación de la masa es más rápida en niños que en adultos, y esto permitiría un enfoque conservador al observar la maduración típica de una lesión de tejidos blandos sospechosa de MO en radiografías convencionales seriadas6.
Se han descrito casos de MOP tras periodos de inmovilización prolongada, como por ejemplo quemaduras, hemofilia, paraplejía, poliomielitis, lesiones craneales y hospitalizaciones prolongadas2. Solo se han reportado 2 casos de niños en edad preescolar, sin patología sistémica previa, con MO tras un periodo prolongado de estancia en la UCI, ambos con afectación bilateral de la musculatura glútea2. Nuestro paciente presentó una afectación unilateral y el periodo de estancia en la UCIN fue menor.
La MO generalmente se presenta como una masa dolorosa de rápido crecimiento sin eritema o calor circundante1. En poblaciones pediátricas sin antecedente traumático, se debe considerar la forma hereditaria de MO (FOP) y la MO secundaria a abuso infantil1,8. La MO progresiva, también llamada FOP o enfermedad de Münchmeyer, es una enfermedad genética grave, extremadamente rara e invalidante1,8,9. Por lo general, aparece de forma esporádica debido a una mutación genética pero también puede heredarse de forma autosómica dominante1,8. La FOP se caracteriza por una osificación heterotópica progresiva del tejido conectivo, que comienza dentro de la primera década de la vida, y una malformación congénita de los dedos gordos que está presente en todas las personas afectas1,8,9. Nuestro paciente no presentó anomalías congénitas externas en los pulgares de los pies, lo que descartó este diagnóstico.
Es fundamental conocer que la MO generalmente tiene 3 etapas de progresión: la etapa temprana, la intermedia y la madura10. Cada una de estas etapas se caracteriza por sus propios hallazgos radiográficos e histológicos y puede conducir a dificultades en la interpretación. Macroscópicamente la MO tiene una estructura similar a un huevo, con una cáscara dura en la periferia y un centro blando. A nivel microscópico, durante la primera semana existe una proliferación de fibroblastos con alto grado de actividad mitótica, hemorragia y necrosis5. En la segunda semana, este núcleo interno se rodea de osteoblastos que producen hueso osteoide inmaduro, a veces con cartílago5. Desde la segunda a la quinta semana, estas 2 zonas quedan delimitadas por una capa externa de hueso laminar maduro, creando el efecto zonal con 3 zonas distintas5. Esta organización zonal es la histopatología característica de la MO en la etapa madura5. A medida que la lesión progresa, madura y se calcifica desde la periferia hasta el centro, siendo este patrón centrípeto la característica diagnóstica más importante para diferenciar la MO de tumores malignos10. Además, la falta de invasión de los tejidos adyacentes y la inclusión de fibras musculares viables en la lesión son útiles en el diagnóstico diferencial8,10. Para descartar con seguridad un proceso neoformativo se realiza una biopsia2. Es importante tomar muestras de todos los elementos de la MO, pues en los estadios iniciales si la biopsia solo se toma de la región central, las células mesenquimales que proliferan rápidamente y tienen una elevada actividad mitótica pueden ser diagnosticadas de malignidad2,5.
De forma análoga a lo que ocurre a nivel histológico, las radiografías simples suelen ser normales en la etapa inicial, por la falta de mineralización en la zona periférica3,10. Durante la etapa intermedia, se puede demostrar una calcificación periférica, mientras que una lesión madura puede aparecer con una calcificación difusa, a medida que el osteoide se mineraliza10. Las calcificaciones aparecen en las radiografías aproximadamente a las 4 semanas8. La evaluación del patrón de calcificación y la afectación ósea ayudan a distinguir la MO de otros procesos.
La prueba de imagen más precisa es la TC, que muestra un patrón suave de osificación normal en el espesor de un músculo, con una clara separación del hueso2. Dependiendo de la etapa de osificación, los elementos del hueso laminar se encuentran en la periferia del músculo osificado con una zona central radiolucente2,7.
Las imágenes de RM pueden ser confusas en los estadios iniciales de la MO, compartiendo muchas características comúnmente encontradas en las neoplasias musculoesqueléticas y los procesos inflamatorios, como lesiones con márgenes poco claros, señales heterogéneas debido a los diferentes elementos celulares de la MO, y presencia de edema difuso en los tejidos circundantes2,6. Las imágenes ponderadas en T1 demuestran una señal desde iso- a hiperintensa, y las ponderadas en T2, una masa heterogénea hiperintensa10. En relación con el edema peritumoral, se ha visto que en niños con tumores musculoesqueléticos aparece en la misma proporción, alrededor de lesiones tanto benignas como malignas6. Aunque es raro, la reacción perióstica puede estar presente en la MO, dependiendo de la ubicación y el desarrollo topográfico de la lesión6. En referencia a esto, se ha clasificado la MO en 3 tipos según su ubicación anatómica: extraósea (en el espesor de un músculo), parostal (evoluciona en la vecindad inmediata o contra un hueso) y perióstica, también conocida como hematoma subperióstico osificante o periostoma6,8. En la MO perióstica y parostal pueden aparecer reacciones periósticas que incluso pueden simular la llamada apariencia de sol naciente típica de sarcomas malignos6. Nuestro paciente presentó tanto edema muscular extenso como una reacción perióstica continua en el fémur; sin embargo, no se encontró destrucción de la cortical, ni anormalidades en la médula ósea, lo que de nuevo hizo improbable el diagnóstico de sarcoma maligno o proceso infeccioso. Sigue siendo una especulación si estos cambios reflejan una respuesta reactiva, cambios inflamatorios subyacentes o simplemente contusión ósea debido a un antecedente traumático.
La gammagrafía ósea es sensible en las primeras fases de la MO, pero no es específica para el diagnóstico2. En nuestro paciente fue útil para excluir lesiones multifocales y el síndrome del niño maltratado.
En pacientes que presentan MO, a menudo se observa una elevación de parámetros inflamatorios en sangre, como VSG, PCR y fosfatasa alcalina1. Nuestro paciente presentó cifras alteradas de PCR en sus primeras horas de vida, sin embargo, no podemos asegurar que fuese la MO el desencadenante.
El diagnóstico diferencial debe incluir neoplasias malignas, así como diversas lesiones benignas y procesos infecciosos. La presencia de edema muscular extenso, así como una reacción perióstica continua en el fémur, pueden sugerir la presencia de un sarcoma maligno, como el osteosarcoma o sarcoma de Ewing; sin embargo, ambos tumores se calcifican del centro a la periferia, al contrario que la MO, están adheridos a la cortical ósea mediante una base amplia, y habitualmente invaden los tejidos adyacentes, presentan anormalidades en la médula ósea y destrucción de la cortical2,5. El osteosarcoma es extremadamente raro en los niños, particularmente sin irradiación previa5. Esta falta de invasión de los tejidos adyacentes y la inclusión de fibras musculares viables en la lesión son útiles en el diagnóstico diferencial histológico de sarcomas extraesqueléticos, parosteales o sinoviales8,10. El rabdomiosarcoma, que representa el 50% de los casos infantiles de sarcoma de tejidos blandos, se observa con mayor frecuencia en la cabeza y el cuello, el tracto genitourinario y las extremidades5. Tiene dos edades pico de incidencia: menores de 5años y mayores de 15años5. En las imágenes, la erosión ósea localizada se asocia frecuentemente con la masa de tejidos blandos y las calcificaciones son poco frecuentes5. El sarcoma sinovial también se debe considerar pero es poco frecuente en niños y se calcifica alrededor del 30%6. Dichas calcificaciones generalmente carecen del borde periférico de osificación que se observa con MO, y suelen mostrar realce intralesional posterior al contraste11,12. El condrosarcoma también es una lesión rara en niños5. Las formas de los tejidos blandos son excepcionales, además, las calcificaciones de tipo condroide presentes dentro de esas lesiones tienen una configuración punteada o de anillo y arco5. Otros procesos menos frecuentes, pero que es importante distinguir de la MO porque pueden tener calcificaciones de tejidos blandos, incluyen la periostitis reactiva y, cuando se implanta en la superficie del hueso, proliferación osteocondromatosa bizarra parostal (o enfermedad de Nora)12. Otra lesión de tejidos blandos que debe diferenciarse es el sarcoma de alto grado indiferenciado pleomórfico óseo o histiocitoma fibroso maligno, ya que puede presentar un componente osteocartilaginoso similar a la MO6.
En la fase aguda de la MO, la apariencia de la RM puede simular un absceso de tejido blando12. Sin embargo, este demuestra clásicamente una apariencia uniforme hiperintensa en secuencias potenciadas en T2, e hipointensa en T1, y un realce periférico tras administración de contraste12. A su vez, la TC con contraste intravenoso demuestra una colección brillante de líquido que realza el borde confirmando la sospecha de absceso12.
Por el fenotipo del paciente y la coexistencia con una displasia del desarrollo de la cadera, y astrágalo vertical congénito bilateral, se planteó la posibilidad de una displasia ósea; sin embargo, no se encontró ninguna entidad nosológica que englobase el conjunto de características clínicas observadas. Se consideró la melorreostosis, una displasia ósea esclerótica benigna rara que sigue una distribución esclerotomal y se sabe que tiene una variante «similar a la miositis»12. La clave para diferenciar la melorreostosis de la MO es la identificación del patrón esclerotomal, lo cual no es característico de la MO12. También se valoró la hiperostosis cortical neonatal o enfermedad de Caffey, una patología ósea inflamatoria rara y autolimitada, casi exclusiva de lactantes, que cursa con fiebre y origina una neoformación perióstica con engrosamiento de la cortical ósea e inflamación de los tejidos adyacentes, pero respeta la epífisis y la metáfisis de los huesos tubulares11; del mismo modo, se contempló el pseudohipoparatiroidismo y sus enfermedades relacionadas, por su frecuente presentación con osificaciones ectópicas subcutáneas o progresivas, pero con alteraciones analíticas ausentes en nuestro caso.
Debido a su baja prevalencia, los principios del tratamiento de la MO se basan en la experiencia empírica más que en la evidencia clínica o experimental, y se fundamentan en el soporte sintomático con fármacos analgésicos y antiinflamatorios no esteroideos, acompañado de reposo relativo, ya que la mayoría de las lesiones se resuelven en un año. Aunque la indometacina se usa con bastante frecuencia en ortopedia para prevenir osificaciones heterotópicas, no ha sido validada para la prevención y/o tratamiento de la MO13. No se han encontrado referencias respecto al uso de los glucocorticoides en esta entidad, pero la literatura no recomienda su uso durante la fase aguda de una lesión del músculo esquelético13. Cuando el diagnóstico clínico y radiológico es incierto, o cuando la lesión causa dolor, bloqueo mecánico de una articulación, o deterioro neurológico, generalmente se realiza la exéresis de la masa3.
Como conclusión cabe destacar que la MO es una entidad rara en niños, y excepcional en recién nacidos. El desafío diagnóstico se fundamenta en diferenciar esta lesión, de tumores malignos óseos y de partes blandas. La sensibilidad de la técnica de imagen elegida está determinada por la etapa de evolución en la que se encuentra. De este modo, aunque existen imágenes características sugestivas de MO, estas no están presentes hasta que, tras una maduración sucesiva de la lesión, se desarrolla el patrón zonal característico. La TC es el estudio que demuestra, de mejor manera y más temprano, el patrón típico de osificación en casos seleccionados en los que se sospecha MO y puede confirmar el diagnóstico. La RM debe realizarse solo si las técnicas de imagen anteriores no han logrado establecer el diagnóstico de MO y todavía se sospecha una neoplasia. El enfoque conservador tiene un papel definido en esta entidad debido a su carácter autolimitado y regresión espontánea. El tratamiento quirúrgico se reserva para aquellas lesiones que provocan restricciones funcionales o deterioro neurológico. El interés del caso radica en la corta edad del paciente, su fenotipo sindrómico asociado a otras alteraciones esqueléticas, un periodo corto de inmovilización, la ausencia de un traumatismo reportado, y un largo periodo de seguimiento. Las características clínicas del paciente permitieron postular una etiología genética, si bien no se estableció un diagnóstico nosológico. Con relación a esto, destacamos la importancia del seguimiento clínico del paciente, pues en ocasiones el fenotipo clínico completa su definición con la edad; pero hasta el momento no han aparecido nuevos signos o síntomas que sugieran una entidad nosológica. Ningún caso de MO en niños menores de 10años descrito en la literatura presentaba patología previa de interés, salvo un caso de parálisis cerebral infantil con MO en ambas caderas14. Se consideraron dos hipótesis etiológicas: en primer lugar, una MO pseudomaligna a consecuencia de la inmovilización durante su estancia en la UCIN, y en segundo lugar, una MO traumática por un traumatismo no reconocido intraparto. Consideramos que, debido a la unilateralidad de la lesión y su corto periodo de inmovilización, es más probable la etiología traumática.
Nivel de evidenciaNivel de evidencia IV.
FinanciaciónTrabajo no financiado.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
