




Las alteraciones hematológicas son comunes en los pacientes con lupus eritematoso sistémico (LES). Pueden expresarse relacionadas con el compromiso de las líneas celulares y con la presencia de alteraciones de la coagulación. El compromiso en la coagulación se asocia con manifestaciones trombóticas. Se han descrito factores de riesgo asociados a trombosis, como la presencia de niveles elevados de homocisteína, déficit adquirido de la proteína S, proteína C y antitrombina. Sin embargo, la diátesis hemorrágica también se ha descrito con menor frecuencia y relacionada con el déficit de factores de la coagulación, secundaria a la presencia de inhibidores. Presentamos 3 pacientes con LES juvenil con manifestaciones hematológicas poco usuales y revisión de la literatura relacionada.
Se concluye que las manifestaciones hematológicas en LES juvenil no solo se relacionan con alteraciones en las líneas celulares. Trombosis vasculares y trastornos hemorrágicos deben sospecharse. El diagnóstico precoz y el tratamiento temprano disminuyen la morbimortalidad relacionada con este tipo de manifestaciones.
Haematological alterations are common in patients with systemic lupus erythematosus (SLE). These haematological manifestations may be expressed related to the involvement of cells affected and coagulation changes. The compromise in coagulation is associated with thrombotic manifestations. Risk factors associated with thrombosis have been described, such as the presence of elevated levels of homocysteine, acquired deficit of protein S, protein C, and antithrombin. However, the haemorrhagic diathesis has also been described at a lower frequency and related to the acquired deficiency of coagulation factors caused by the development of autoantibodies directed against coagulation factors. The cases are presented of 3 patients with juvenile SLE with unusual haematological manifestations, as well as a review of the literature in relation to them.
The haematological manifestations in juvenile SLE are not only related to alterations in cell lines, vascular thrombosis and bleeding disorders should also be suspected. Early diagnosis and treatment reduces morbidity and mortality related to this type of manifestations.
El lupus eritematoso sistémico (LES) es considerado el prototipo de enfermedad autoinmune asociado a un componente de autoinflamación. Se caracteriza por presentar compromiso multisistémico y manifestaciones clínicas heterogéneas en el curso de la enfermedad1,2.
El Colegio Americano de Reumatología (ACR) desarrolló los primeros criterios de clasificación en 1971, los cuales fueron modificados en 1982. Posteriormente, en el 2012 las Clínicas de Colaboración Internacional de Lupus Sistémico (Systemic Lupus International Collaborating Clinics, SLICC) establecieron un nuevo grupo de criterios, los cuales incluyen 11 criterios clínicos y 6 inmunológicos. Se requiere la presencia de 4 criterios dentro de los cuales debe haber uno clínico y uno inmunológico o una biopsia renal con hallazgos compatibles con nefritis lúpica asociados a la presencia de anticuerpos antinucleares (ANA) o anticuerpos anti-dsADN3. Los criterios ACR tienen una sensibilidad de 76,6% y especificidad de 93,4% a diferencia de los criterios SLICC, los cuales son más sensibles que específicos: 98,7% y 85,3%, respectivamente3. Hartman et al. publicaron un metaanálisis, en el cual, basados en la sensibilidad y especificidad para la población pediátrica y con el fin de evitar falsos positivos, recomiendan el uso a reumatólogos pediatras de los criterios del ACR4.
La incidencia y prevalencia de la enfermedad son variables. Se ha estimado una prevalencia de 3,3-8,8 por cada 100.000 personas en edad pediátrica y una incidencia de 0,3-0,9 por cada 100.000 personas/año. La edad de inicio es variable, si bien es más frecuente entre los 12 y 16 años, con predominio en el género femenino (4,5-5:1)1,5,6. El LES de inicio en edad juvenil (LESj) representa del 10-25% de todos los casos de LES1-3.
En cuanto a las manifestaciones clínicas del LESj, el compromiso hematológico es un hallazgo común, su frecuencia oscila en diferentes series entre el 50 y el 100% de los pacientes y puede comprometer la línea roja, blanca, plaquetas o el sistema de coagulación2,5.
De las alteraciones hematológicas, las más frecuentes son las alteraciones en las líneas celulares. En primer lugar, la anemia está presente en el 80-90%, su etiología en orden decreciente es anemia por enfermedad crónica, anemia por deficiencia de hierro y anemia hemolítica1-3. En esta última, descrita en aproximadamente el 10-15%, es de resaltar que el 30-40% de los pacientes presentan prueba de Coombs directa positiva sin otros hallazgos sugestivos de hemólisis3.
Entre las alteraciones de la línea celular blanca, la más frecuente es la leucocitopenia: se presenta entre el 46 y el 64% de los pacientes con LESj. Es posible encontrar neutropenia y linfopenia. Es más prevalente esta última y es considerada un marcador de actividad de la enfermedad1-3. Por último, la trombocitopenia está presente en el 7-30% de los pacientes y aproximadamente en el 15% de los casos al inicio de la enfermedad en edad pediátrica3.
El segundo escenario clínico en cuanto a las manifestaciones hematológicas en LESj son las alteraciones en la coagulación, que pueden ser trombóticas o hemorrágicas; son más frecuentes las trombóticas. Si bien su frecuencia no es tan alta como las alteraciones en las líneas celulares, se asocian a alta morbimortalidad1-5. En el presente artículo se presentan 3 pacientes con LESj con manifestaciones hematológicas poco usuales y revisión de la literatura relacionada.
Reporte de casosCaso 1Paciente masculino de 15 años de edad, con diagnóstico a los 13 años de síndrome de anticuerpos antifosfolípidos (SAF), que cumplía criterios de trombosis venosa profunda a nivel de poplítea derecha, con anticoagulante lúpico positivo y anticardiolipinas IgG e IgM positivas. Recibió manejo anticoagulante con heparinas de bajo peso molecular y cloroquina durante 8 meses, con abandono de tratamiento.
Ingresó a nuestro servicio por fiebre persistente y lesiones vasculíticas en miembros inferiores de un mes de evolución. Al examen físico, se encontró hipertensión arterial estadio ii, adenomegalias cervicales y axilares, fenómeno de Raynaud severo con lesiones iniciales de ulceración en pulpejos de dedos de manos y pies, y edema en miembros inferiores. En los paraclínicos iniciales se evidenció leucocitopenia, linfopenia, anemia normocítica normocrómica, prueba de Coombs directa negativa, reticulocitos normales, ANA título 1/160, anti-ADN título 1/320, anti-RO y anti-La positivos, anticoagulante lúpico e IgG e IgM anticardiolipina positivos, hipocomplementemia de C3 y C4, proteinuria en rango nefrótico y ecografía doppler venosa de miembros inferiores con trombosis venosa profunda crónica parcialmente recanalizada, que comprometía vena poplítea derecha sin hallazgos sugestivos de trombos agudos ni compromiso arterial.
Se consideró diagnóstico de LES y SAF. Se inició manejo inmunosupresor con pulsos de metilprednisolona y ciclofosfamida asociado a medidas de protección al frío, ácido acetil salicílico, antimaláricos, manejo antihipertensivo y nefroprotección con inhibidores de la enzima convertidora de angiotensina y anticoagulación con enoxaparina ajustada a la función renal.
Durante el curso de la hospitalización, el paciente presentó deterioro progresivo de la perfusión distal de dedos de manos y pies, requirió manejo en UCIP. Se adicionó al manejo antagonistas de receptores de angiotensina (ARA II), nitroglicerina tópica y pentoxifilina, sin haber obtenido respuesta. Por el contrario, hubo progresión de la lesión isquémica, por lo que se inició plasmaféresis y se realizó bloqueo simpático con mejoría moderada de la perfusión en el pie derecho. No obstante, se presentó deterioro de las lesiones en mano izquierda, cianosis marcada con áreas de isquemia y necrosis en dedos de la mano derecha y pie derecho, por lo que se adicionó al manejo sildenafil y bosentán, lo que permitió la estabilización del cuadro clínico. Presentó mejoría de la perfusión en algunas áreas y exéresis espontánea del tercer dedo del pie derecho, sin requerimiento de manejo quirúrgico de las otras zonas de necrosis. La adecuada evolución permitió el descenso progresivo de bosentán. Actualmente, el paciente se encuentra en manejo con tratamiento anticoagulante, ácido acetil salicílico, antihipertensivo, antimalárico y ciclofosfamida, y presenta mejoría de las cifras tensionales y de la proteinuria.
Durante el seguimiento, se solicitaron estudios para descartar otros factores de trombofilia adicionales a SAF. Dentro de los cuales se obtuvo: mutación homocigota del gen de metilentetrahidrofolato reductasa (MTHFR) C. 677C>T (P. A222V), mutación del gen protrombina negativo, proteína C de coagulación (131% dentro del rango de referencia), factor V (FV, 85%) y factor VIII (FVIII, 51%), normales. Por lo que se considera factor de riesgo protrombótico adicional a los anticuerpos antifosfolípidos dado por la presencia de mutación en el gen MTHFR.
Caso 2Paciente femenina de 16 años de edad, previamente sana, sin antecedentes familiares o personales relevantes. Ingresó por manifestaciones clínicas de un mes de evolución caracterizadas por síntomas respiratorios, disnea de medianos esfuerzos, dolor torácico y tos húmeda, asociados a síntomas constitucionales (astenia, adinamia y fiebre), poliartritis y edema en miembros inferiores. Durante la valoración inicial en urgencias, se encontró con hipertensión arterial, taquicardia, sin fiebre, sin dificultad respiratoria, edema facial y en miembros inferiores, sin lesiones en cavidad oral, sin linfadenopatías ni megalias y sin lesiones en piel. En los paraclínicos iniciales se evidenció leucocitopenia, anemia normocítica normocrómica, prueba de Coombs directa negativa, trombocitopenia, elevación de nitrogenados, proteinuria en rango nefrótico, hematuria y esquistocitos en el extendido de sangre periférica.
En el periodo mediato al ingreso, presentó deterioro clínico dado por hemoptisis, derrame pleural y pericárdico, y dificultad respiratoria que conllevó falla respiratoria hipoxémica. Se estableció impresión diagnóstica de LES considerando la presencia de síntomas constitucionales, compromiso articular, hematológico (leucocitopenia, anemia hemolítica, trombocitopenia), renal (falla renal aguda, proteinuria, hematuria), inmunológico (anti-ADN título 1/640, ANA título 1/320 patrón homogéneo, anti-La y anti-Ro positivos e hipocomplementemia), serositis pulmonar (hemorragia alveolar difusa), microangiopatía trombótica (esquistocitosis, anemia hemolítica, Coombs negativo, LDH elevada, hiperbilirrubinemia a expensas de la bilirrubina indirecta, haptoglobina consumida y trombocitopenia).
La paciente fue trasladada a UCIP, donde se inició soporte ventilatorio, hemodinámico y renal (hemodiálisis) y, para su enfermedad de base, pulsos de metilprednisolona asociados a ciclofosfamida intravenosa (2 dosis). Tras la administración de la segunda dosis, se evidenció leucocitopenia y trombocitopenia severa y se decidió inicio de inmunoglobulina por refractariedad, de la cual recibió 5 dosis, sin obtener control del sangrado pulmonar, por lo que se prescribió rituximab, con adecuada tolerancia y control del sangrado inicialmente. Después presentó deterioro de los síntomas y trombocitopenia severa por lo que se inició plasmaféresis durante 5 sesiones.
Adicionalmente, y teniendo en cuenta la manifestación de microangiopatía trombótica con ADAMTS 13 sin evidencia de deficiencia en la actividad enzimática, se consideró síndrome hemolítico urémico atípico y se inició manejo con eculizumab, el cual permitió estabilizar la función renal y el recuento plaquetario de forma transitoria.
Durante el curso de la hospitalización se documentó infección por citomegalovirus, por lo que se aplazó la administración de la segunda dosis de rituximab hasta 60 días después de la primera. Con las intervenciones realizadas hubo control parcial del sangrado y la trombocitopenia, mejoría de la función renal, con reagudizaciones frecuentes. Se amplió el estudio y se descartaron alteraciones de la coagulación asociadas (cianocobalamina 341 pcg/mcl (valor de referencia [VR] 189-883 pcg/mcl), inhibidor del FIX 0 UB, factor von Willebrand (FvW, 203%; VR 50-160%), inhibidor del FVIII 0 UB, cofactor de ristocetina 107,3% (VR 50-150%), FVII (111%). Sin embargo, por refractariedad de hemorragia pulmonar con sangrado que amenaza la vida, se inició suplencia con FVIIa intravenoso, con mejoría clínica inicial.
A los 3 meses de su ingreso, debido a su evolución tórpida y deterioro del estado clínico, se realizó biopsia pulmonar, que evidenció ensanchamiento del intersticio dado por edema, congestión vascular, focos de capilaritis y hemosiderosis. Así mismo, se realizó aspirado de médula ósea, que evidenció hemofagocitos, sin hallazgos sugestivos de malignidad. Además de los hallazgos histológicos, se documentó hiperferritinemia (3.982 ng/ml), hipertrigliceridemia y anemia, por lo que se consideró un nuevo escenario en la paciente, consistente en síndrome de activación macrofágica (SAM), de posible etiología multifactorial. Se inició manejo de acuerdo con las guías de tratamiento de la Sociedad Internacional del Histiocito del 2004 con dexametasona, ciclosporina y etopósido, sin obtener respuesta clínica adecuada, al contrario, con deterioro clínico progresivo. Presentó episodio de sangrado pulmonar masivo a pesar de manejo inmunosupresor y suplencia diaria de FVIIa, que llevó al fallecimiento de la paciente a los 3 meses de la hospitalización.
Caso 3Paciente femenina de 8 años de edad, quien ingresó a urgencias de la institución por clínica que inició 12 meses previos a la consulta, consistente en edema intermitente en manos, rigidez matinal y dolor de características inflamatorias, los cuales se agudizaron 2 meses previos a su ingreso. Asociados y de igual tiempo de evolución, presentaba equimosis en miembros inferiores y sangrado gingival. Durante la valoración inicial se encontró, al examen físico, eritema malar, epistaxis, poliartritis (muñecas bilaterales, edema en articulación interfalángica proximal del 4.° dedo de la mano derecha y en el 2.°, 3.° y 4.° dedos de la mano izquierda), equimosis perimaleolar en el cuello del pie derecho y en la rodilla ipsolateral, así como periartritis del tobillo derecho, sin evidenciarse adenomegalias ni visceromegalias. En los paraclínicos iniciales, se observó hemograma sin citopenias, reactantes de fase aguda (VSG y PCR) elevados, función renal y hepática dentro de parámetros normales, uroanálisis con adecuada acidificación y concentración urinaria sin proteinuria, hematuria microscópica, hipocomplementemia de C3 y C4, tiempos de coagulación prolongados (PT 22,3/14,8; PTTa 59,5/29,9). Los valores del perfil de autoinmunidad fueron ANA positivos (título 1/2.560), anti-dsADN positivo (título 1/160), anticuerpos antiantígenos extractables negativo. Se consideró LES como impresión diagnóstica teniendo en cuenta el compromiso cutáneo, articular, renal (hematuria) e inmunológico (ANA, anti-ADN e hipocomplementemia). Debido a la presencia de tiempos de coagulación prolongados y evidencia de sangrado activo, también se consideró un posible trastorno de coagulación adquirido. Se hospitalizó a la paciente para complementar estudios e iniciar manejo médico con pulsos de metilprednisolona por 3 días y antimalárico.
Durante la hospitalización, presentó evolución clínica satisfactoria, con mejoría parcial de compromiso articular y sin evidencia de nuevos sangrados. En los paraclínicos obtenidos durante su hospitalización, se evidenció anticoagulante lúpico presuntivo positivo, LDH normal, reticulocitos elevados, extendido de sangre periférica con anisocitosis sin otras alteraciones y prueba de Coombs directa negativa, radiografía de tórax sin evidencia de serositis y ecografía abdominal normal. Se adicionó al manejo azatioprina y se dio egreso con control ambulatorio y valoración de reporte de cuantificación de factores de coagulación, los cuales se encontraban pendientes para esa fecha.
En el seguimiento presentó evolución satisfactoria, con paraclínicos de control con anticardiolipinas negativas, FVIII (17,8%; VR 50-150%) y FIX (3,8%; VR 50-150%) disminuidos, proteína S, FV de Leiden, proteína C y tiempos de coagulación normales. Hematología consideró hemofilia adquirida y, debido a la respuesta inicial adecuada al manejo inmunosupresor, se decidió continuar con el mismo manejo instaurado. Durante los controles ambulatorios, se evidenció mejoría progresiva de niveles de factores de coagulación: FVIII (19%) y FIX (24%), hasta alcanzar la normalidad durante último control (FVIII: 169% y FIX: 95%), por lo que se consideró hemofilia adquirida en remisión.
Revisión de la literaturaEn la literatura se han reportado alteraciones en el sistema de la coagulación en pacientes con LES, consecuencia del déficit de los factores de coagulación secundario a la presencia de anticuerpos inhibitorios o anticuerpos protrombóticos. En los 3 casos anteriores se presentan diferentes espectros de la enfermedad. En el primer caso, hemorragia pulmonar masiva; en el segundo caso, trombofilia por presencia de anticuerpos antifosfolípidos asociada a la mutación homocigota en el gen de la MTHFR y en el tercer caso, un paciente con hemofilia adquirida. Teniendo en cuenta que los pacientes presentaron manifestaciones relacionadas con alteraciones en la coagulación, se describirán a continuación los mecanismos fisiológicos de la coagulación, con el fin de entender los mecanismos patológicos en cada paciente.
La hemostasia es el mecanismo que permite proteger al organismo de la pérdida de sangre ante cualquier lesión que se produzca en los vasos sanguíneos. Su objetivo es activar diferentes procesos fisiológicos que conducen a 1) la formación del trombo hemostático, 2) la reparación del daño y 3) la disolución del coágulo, lo cual se manifiesta clínicamente con el control de la hemorragia7,8.
Décadas atrás se describió la cascada de la coagulación, que comprendía una vía clásica con 2vías de activación (intrínseca y extrínseca) y que convergían en una vía común9. En la actualidad se conoce que la hemostasia no es posible sin las células que expresan el factor tisular: plaquetas, células endoteliales, monocitos y fibroblastos, así como otras sustancias procoagulantes y anticoagulantes8,9.
El modelo actual de la coagulación es un modelo celular. Este modelo se desencadena en la membrana de células que expresan el factor tisular y se divide en 3 fases8,9.
- 1)
Fase de iniciación: cuando la vasculatura es lesionada y las células endoteliales son expuestas, se favorece la liberación del factor tisular inactivo en la superficie celular en pequeñas cantidades. Este se une al FVII (FVII) que actúa como cofactor e induce su activación (FVIIa). De esta forma, se constituye el complejo factor tisular/FVIIa, que activa directamente al factor X (FXa) e indirectamente al factor IX (FIXa). Esto permite que el FXa se una al FV activado (FVa) para formar un complejo protrombinasa en la superficie y convertir la protrombina en trombina, pero en cantidades insuficientes para la formación de fibrina7-9.
- 2)
Fase de amplificación: la trombina previamente acumulada activa las plaquetas adheridas por un receptor específico (glicoproteína Ia/IIa) y el FvW. De esta forma la trombina activa el FV, amplificando la actividad protrombinasa y activando el FVIII (FVIIIa), el cual a su vez actúa como cofactor del FIXa para mantener la generación del FXa. En esta fase, la plaqueta tiene en su superficie factores activados junto con el FvW y se lleva a cabo la activación de los anticoagulantes naturales: TFPI (inhibidor del complejo TF/FVIIa), antitrombina y proteína C, importantes en la regulación del efecto procoagulante7-9.
- 3)
Fase de propagación: en la superficie plaquetaria el FXIa activa el FIX. El FIXa se une al FVIIIa, establece el complejo tenasa (FIXa+FVIIIa+Ca+2), cataliza la activación de FX e incrementa la conformación del complejo FXa/FVa+Ca+2. Este complejo cataliza la conversión de trombina para la formación de fibrina. La fibrina activa el FXIII (factor estabilizador de fibrina), el cual forma enlaces covalentes entre cadenas de fibrina para la formación y estabilización del coágulo7-9.
En LES las manifestaciones trombóticas tienen una prevalencia del 10-50%, según los factores de riesgo presentes en cada paciente7. Se relacionan principalmente con la positividad de anticuerpos antifosfolípidos (aPL). La prevalencia de anticoagulante lúpico es del 20-30%, de beta 2 glicoproteína del 40% y de anticuerpos anticardiolipina del 42%9. El 50-80% de los pacientes con aPL en LES cumplen criterios de clasificación para SAF, con incremento de la morbilidad, ya que aumenta el riesgo de trombosis de 20 a 30 veces más que los pacientes sin aPL3. Cervera et al. observaron, en su cohorte de 1.000 pacientes con LES, que el 9,2% de los pacientes presentaron el antecedente de trombosis durante la inclusión al estudio y que el 1,8% de los pacientes incluidos fallecieron como consecuencia de episodios trombóticos, los cuales estuvieron asociados a la presencia de aPL10. A continuación se describirán los factores de riesgo asociados a trombosis y se hablará con más detalle del SAF.
Los niveles elevados de homocisteína son considerados como un factor de riesgo independiente para ateroesclerosis, trombosis arterial y venosa11. La hiperhomocisteinemia puede ser secundaria a deficiencia de la vitamina B12, vitamina B6, ácido fólico, falla renal crónica, hipotiroidismo o mutaciones en el gen de la MTHFR11,12. Sallai et al. describieron que aproximadamente el 15% de su cohorte de 105 pacientes con LES presentaron niveles elevados de homocisteína y que el 27,3% de los pacientes con hiperhomocisteinemia desarrollaron trombosis comparados con el 16,9% que no presentaron trombosis13.
La MTHFR es una enzima que interviene en el metabolismo de la homocisteína, aminoácido sulfurado producto del metabolismo de la metionina que procede de las proteínas de la dieta. El gen de la MTHFR se localiza en el cromosoma 1p36.2 y la mutación C677T consiste en la sustitución de una citosina por una timina en el nucleótido 677, lo que origina la sustitución de una alanina por una valina en la posición 223. Este cambio genera una variante de MTHFR termolábil que se caracteriza por la disminución del 50% en su actividad a 37°C, lo que condiciona la presencia de niveles de homocisteinemia elevados que se relacionan con la predisposición a trombosis12,14,15. Dentro de los mecanismos descritos por los cuales la hiperhomocisteinemia favorece los episodios trombóticos, se encuentran el incremento en la proliferación de las células musculares, la inhibición de la síntesis de ADN de células endoteliales, el aumento de la respuesta vasoconstrictora, la reducción de la expresión de la trombomodulina, el incremento en la expresión de factor tisular, la inhibición de la expresión de heparansulfato y la liberación de óxido nítrico y prostaciclinas, y la reducción de la unión del activador tisular del plasminógeno a su receptor endotelial12,13.
La Academia Americana del Corazón (AHA) considera que los resultados de los estudios relacionados con la presencia de mutaciones en el gen de la MTHFR han sido contradictorios y que su importancia clínica en el riesgo de eventos trombóticos es incierta15. No obstante, algunos autores la consideran como factor de riesgo adicional a la presencia de aPL en el desarrollo de fenómenos trombóticos, como se observó en el paciente del caso 116-18. Giannelou et al. identificaron en una cohorte de 150 pacientes con LES que tanto la hiperhomocisteinemia (OR 5,8; IC 95%: 1,0-35,8) como el genotipo MTHFR 677TT (OR 5,2; IC 95%: 1,1-24,0) actuaron como factores independientes para la formación de placa ateroesclerótica. Además, que el genotipo MTHFR confirió un mayor riesgo de engrosamiento de la pared arterial (OR 4,9; IC 95%: 1,2-20,6) sin que se reprodujera este hallazgo con la hiperhomocisteinemia. Estos hallazgos señalan que influencias genéticas como la variante MTHFR 677TT aumenta la carga de enfermedad ateroesclerótica que caracteriza al LES19. En la actualidad no existen estudios que indiquen la frecuencia ni el riesgo de trombosis en pacientes con LESj relacionado con la presencia de mutaciones en este gen.
El déficit adquirido de la proteína S, proteína C o antitrombina (AT III) son infrecuentes. No obstante, su presencia favorece el riesgo de trombosis. El déficit adquirido de proteína C ha sido reportado hasta en el 50% de los pacientes con anticoagulante lúpico9,20. Además de esto, en pacientes con síndrome nefrótico puede encontrarse con frecuencia el déficit adquirido de AT III8. En conclusión, los defectos congénitos que condicionan las trombofilias no se ven incrementados en pacientes con LES, pero, como se había descrito previamente, su presencia se asocia a mayor riesgo de eventos trombóticos3,19,20.
Síndrome antifosfolípidoEl SAF es una enfermedad autoinmune sistémica que se caracteriza por la presencia de eventos tromboembólicos, morbilidad en el embarazo y títulos elevados de aPL. El 50% de los pacientes presentan la enfermedad aislada, es decir, no se encuentra asociada a otra enfermedad autoinmune. No obstante, se ha descrito que hasta el 50% la presentan asociada a otra enfermedad autoinmune: entre el 40 y el 80%, asociada a LES21-23.
Los aPL son anticuerpos que pueden ser de los 3 idiotipos (IgG, IgM o IgA), los cuales están dirigidos contra los complejos proteicos (beta 2 glicoproteína, protrombina, anexina V, proteína C, proteína S) o los fosfolípidos de las membranas celulares (cardiolipina, fosfatidilserina, fosfatidiletanolamina, fosfatidilinositol). Se ha descrito su principal interacción con la beta 2 glicoproteína y la anexina V21-23.
Los efectos que generan en las células se relacionan con la activación y agregación plaquetaria, activación de monocitos, liberación de citocinas proinflamatorias como IL1, IL6 e IL8 e incremento en la expresión de moléculas de adhesión (ICAM, VCAM y E selectina). En el endotelio, inducen la expresión del factor tisular y moléculas de adhesión y su efecto en la coagulación se relaciona con la formación del coágulo de fibrina, disminución de la fibrinólisis al inhibir la trombina, proteína C, plasminógeno y plasmina. Los efectos descritos conducen al incremento de la lesión tisular, lo cual favorece el desarrollo de las manifestaciones trombóticas21,22.
Avcin et al. publicaron una cohorte multicéntrica con 121 pacientes con SAF de inicio en edad pediátrica. El 49,5% de los pacientes no tenía otra enfermedad autoinmune y el 38% de los pacientes tenía asociado LESj. Se observaron las manifestaciones trombóticas en el 38% de los pacientes: la trombosis venosa fue la más frecuente (26%) y con menor frecuencia, la arterial (6%), y las manifestaciones no trombóticas en el 40% de los pacientes. El compromiso hematológico se describió en el 38%, principalmente relacionado con la anemia hemolítica con prueba de Coombs positiva, trombocitopenia de leve a moderada, síndrome de Evans (trombocitopenia asociada a anemia hemolítica inmune) y leucocitopenia. Las manifestaciones dermatológicas se registraron en el 18% de los pacientes, relacionadas con livedo reticularis, fenómeno de Raynaud, úlceras cutáneas y urticaria crónica y el 16% desarrollaron manifestaciones neurológicas no trombóticas (cefalea migrañosa, desórdenes del movimiento, epilepsia y trastornos del afecto con menor frecuencia)24. Estos datos concuerdan con lo descrito por otros autores25,26.
Se han validado en población adulta los criterios de clasificación de Sapporo para establecer el diagnóstico de SAF, dentro de los cuales se encuentra la morbilidad en el embarazo como criterio clínico. Sin embargo, para la población pediátrica no es considerado como criterio de clasificación21. Por otra parte, en las recomendaciones publicadas recientemente en las guías EULAR, se describe la necesidad de validar unos criterios de clasificación en la población pediátrica en los que se incluyan manifestaciones no trombóticas, ya que, como se describió previamente, son manifestaciones clínicas frecuentes en este grupo etario25 (tabla 1)22.
Criterios de clasificación Sapporo revisados para SAF
| Criterios de Sapporo revisados en pacientes pediátricos | |
|---|---|
| Criterios clínicos | Trombosis vascular → Uno o más episodios de trombosis de pequeños vasos, arterial o venosa confirmada por imágenes o patología (presencia de trombosis con evidencia de inflamación en la pared del vaso) |
| Criterios laboratorio | Anticoagulante lúpico → (+) en 2 o más ocasiones, diferencia de 12 semanasAnticuerpos IgG/IgM anticardiolipina →En suero o plasma en títulos medios o altos en 2 o más ocasiones, diferencia 12 semanas (ELISA)Anticuerpos IgG/IgM beta 2 glicoproteína →- En suero o plasma en títulos altos en 2 o más ocasiones, diferencia 12 semanas (ELISA). |
Se establece SAF con un parámetro clínico y uno paraclínico.
Fuente: Tomado de Petty et al.22.
Aunque tradicionalmente se ha descrito la asociación de aPL con episodios trombóticos, estos anticuerpos también pueden estar dirigidos contra la protrombina y acelerar su depuración por el sistema retículo endotelial, asociándose de esta forma a diátesis hemorrágica25.
El hallazgo de aPL es esencial para establecer el diagnóstico. El anticoagulante lúpico es más específico para SAF, mientras que los anticuerpos anticardiolipina son más sensibles. La especificidad de estos últimos puede incrementarse ante la presencia de títulos altos de idiotipo IgG de anticuerpos anticardiolipina26. Aunque la positividad se considere como predictor de riesgo de trombosis en pacientes con LES o SAF, no se ha descrito una asociación definitiva entre las manifestaciones clínicas y un aPL específico26,27. Driest et al. publicaron un estudio que incluyó a 979 pacientes con manifestaciones trombóticas en LESj, en el que encontraron que la positividad de los aPL fue estadísticamente significativa (p=0,0052). Estos hallazgos los llevaron a definir los aPL como factor de riesgo para trombosis en pacientes con LESj18.
Groot et al. publicaron las recomendaciones de manejo en pacientes con SAF de inicio en edad juvenil. En pacientes con LES y ante la presencia de aPL, se debe considerar para la prevención primaria la adición de un antiagregante, idealmente ácido acetil salicílico. Sin embargo, también debe tenerse presente que los antimaláricos tienen efecto antiagregante. La recomendación ideal es el uso concomitante de ácido acetil salicílico y antimalárico. Ante episodios trombóticos venosos y aPL persistentemente positivos, se indica terapia anticoagulante a largo plazo con un objetivo terapéutico de INR entre 2 y 3. En caso de evento trombótico arterial y aPL persistentemente positivos, se indica terapia anticoagulante a largo plazo o terapia anticoagulante y antiagregante combinada e INR ideal entre 2 y 3. Los pacientes con síndrome antifosfolípido catastrófico no solo requieren terapia anticoagulante, sino que además se les indica el uso inmediato de dosis altas de glucocorticoides, plasmaféresis y, en casos refractarios, se les adiciona gammaglobulina, rituximab u otro inmunosupresor28.
Manifestaciones hemorrágicas y lupus eritematoso sistémicoEn la literatura se han publicado reportes de casos relacionados con diversas manifestaciones hemorrágicas en pacientes con LES. En nuestro caso, el paciente número 2 presentó hemorragia alveolar y la paciente número 3 presentó hemofilia adquirida.
Hemorragia alveolar y lupus eritematoso sistémicoLa hemorragia alveolar en pacientes con LES es una manifestación clínica rara y potencialmente fatal29. Los mecanismos fisiopatológicos no están completamente definidos. Se ha implicado la disrupción de la pared de los vasos sanguíneos por una vía inmunológicamente mediada, el depósito de complejos inmunes y complemento, las infecciones pulmonares y las coagulopatías29-31.
Existen pocos reportes de casos que han permitido estimar una frecuencia aproximada entre el 0,2 y el 5,4% en pacientes con LES, que se presenta con mayor frecuencia en población juvenil que en adultos y en el género femenino29,32. Badsha et al. describieron que usualmente se presenta en un tiempo medio de 3 años posterior al inicio del LES32. Sin embargo, otros autores describen que en el 30% de los casos la hemorragia alveolar es la primera manifestación de la enfermedad29.
Las manifestaciones clínicas incluyen disnea de inicio súbito, dificultad respiratoria que progresa a falla respiratoria hipoxémica, tos, hemoptisis y, a nivel paraclínico, se observa descenso de los niveles de hemoglobina y evidencia radiológica de infiltrados intersticiales o alveolares difusos29-32. La tasa de mortalidad registrada en la literatura se encuentra entre el 30 y el 90% de los pacientes29.
Araujo et al. publicaron un estudio retrospectivo que incluyó a 13 pacientes con LESj y hemorragia alveolar. Los pacientes incluidos tuvieron una edad media al inicio del LES de 12,7±4,2 años y una edad media al inicio de la hemorragia alveolar de 15,3±2,7 años y el 77% de los pacientes eran mujeres. Las manifestaciones clínicas reportadas fueron hemoptisis, disnea, tos e hipoxemia en el 100% de los pacientes, un descenso significativo de niveles de hemoglobina en el 36,4% (disminución media de 2,9±0,9g/dl) y una tasa de mortalidad del 69% de los pacientes (todas las muertes fueron secundarias a falla respiratoria desencadenada por la hemorragia alveolar, el 50% presentaron infecciones asociadas y un paciente falleció a consecuencia de SAM)29. Estos hallazgos coinciden con lo descrito en el caso del paciente número 2, quien presentó una infección por citomegalovirus que retardó la continuidad del manejo inmunosupresor, presentó pobre respuesta del sangrado alveolar al tratamiento establecido y el desarrollo del SAM que llevaron, finalmente, al fallecimiento del paciente.
El tratamiento farmacológico indicado incluye dosis altas de esteroides, agentes citotóxicos, inmunoglobulina intravenosa y plasmaféresis29,32. En casos refractarios, está indicada la administración de FVIIa intravenoso o local31. El reconocimiento precoz mejora el pronóstico y la posibilidad de respuesta al tratamiento29-32.
Hemofilia adquirida y lupus eritematoso sistémicoLa presencia de manifestaciones hemorrágicas secundaria a autoanticuerpos circulantes (inhibidores) que neutralizan parcial o completamente la actividad o aceleran la destrucción de factores de la coagulación específicos definen la hemofilia adquirida33-37. Se estima que aproximadamente el 16% de los casos registrados corresponden a enfermedades de origen autoinmune, de los que el LES es la más frecuente34.
Los inhibidores dirigidos contra el FVIII son los más frecuentemente encontrados, de aquí el nombre de hemofilia A adquirida, pero se pueden identificar inhibidores para cualquier otro factor de la coagulación30,35.
La incidencia reportada se estima en aproximadamente 0,2-1 por millón de pacientes/año. En el 50% de los casos es secundaria a condiciones como posparto, malignidad y enfermedades autoinmunes, en donde se destaca el LES, artritis reumatoide y arteritis de la temporal34-36. Algunos autores han reportado que la presencia de inhibidores en LES se encuentran en aproximadamente el 5,6% (10 de 215 pacientes). Sin embargo, la incidencia exacta es desconocida, debido a que con frecuencia es una condición subdiagnosticada34-36. La presencia de inhibidores adquiridos se encuentra con mayor frecuencia en pacientes adultos. En menores de 16 años se estima una incidencia de 0,045 por millón de pacientes34.
La manifestación clínica principal es el sangrado (descrita en el 89% de los pacientes), que puede ser espontáneo o presentarse posterior a un trauma, cirugía o posparto y cuya severidad es variable. Las manifestaciones hemorrágicas reportadas son la epistaxis, sangrado gingival, equimosis, petequias e incluso hemorragias severas y potencialmente fatales en el tracto respiratorio, tracto gastrointestinal y sistema nervioso central. Entre el 5 y el 10% de los casos descritos se presentan con sangrado severo, por lo que se considera una condición con alto riesgo de morbimortalidad34,35. A diferencia de la hemofilia congénita, la hemartrosis es infrecuente34. Por el contrario, una pequeña proporción de pacientes no presenta manifestaciones hemorrágicas y en estos casos la prolongación en los tiempos de coagulación (PTTa o PT) es la única alteración sugestiva de la enfermedad34,38,39.
Es importante sospechar el diagnóstico en pacientes con manifestaciones hemorrágicas, sin antecedentes personales ni familiares relevantes para condiciones hemorrágicas y con la presencia de tiempos de coagulación anormales. Al realizar las pruebas de coagulación es posible encontrar varios escenarios: a) Un PTTa anormal con PT normal y, al realizar la prueba de PTTa corregido, en la cual se mezcla el plasma del paciente con plasma normal, si el PTTa corrige indica una deficiencia de factores de la vía intrínseca de coagulación (XII, XI, IX, o VIII), pero en aquellos pacientes en los que persiste prolongado indica presencia de inhibidores a alguno de los factores de coagulación o la presencia de anticuerpos antifosfolípidos. b) En PT y PTTa anormal con una prueba de mezclas que no corrige, se debe sospechar la presencia de inhibidores dirigidos con los FV, FII y FX35-40.
Una vez se sospeche el diagnóstico, se debe realizar una medición cuantitativa de inhibidores (en el caso de la hemofilia A adquirida, inhibidores específicos dirigidos contra el FVIII). Además, es importante tener presente que los niveles de los inhibidores no tienen relación directa con la severidad del sangrado ni con la mortalidad34. También es posible encontrar niveles bajos del FVIII, y otras pruebas de hemostasia como conteo plaquetario suelen ser normales35. Estas alteraciones coinciden con los hallazgos descritos en la paciente del caso clínico número 3 (PT yPTTa prolongados asociados a niveles disminuidos del FVIII y FIX), lo que permitió establecer el diagnóstico de hemofilia adquirida.
Los inhibidores se cuantifican mediante la prueba «Bethesda» en la que se mezcla el plasma normal (como fuente de FVIII o FIX) con plasma del paciente sin diluir y esta mezcla se incuba durante 2 h a 37°C. Una unidad Bethesda se define como la cantidad que destruye la mitad del factor en esa muestra corregida38. Los aloanticuerpos son policlonales de tipo IgG1 e IgG4; estos se producen por pérdida de tolerancia inmune que regula la respuesta al FVIII34,35.
El tratamiento consiste en controlar el sangrado y eliminar los inhibidores. Para el control de sangrado se recomiendan el uso del FVIII humano o porcino, concentrado de protrombina activado (complejo coagulante antiinhibidor) o FVIIa recombinante no humano en caso de sangrado severo. En pacientes con sangrado leve no es necesaria la aplicación de concentrados de factores de coagulación35,37.
La eliminación de los inhibidores requiere una terapia inmunosupresora a largo plazo con corticoides, agentes citotóxicos como ciclosporina y ciclofosfamida o terapia combinada; de igual forma también están indicadas, según la severidad del sangrado, inmunoglobulina intravenosa o plasmaféresis35-38. El régimen actual recomendado como primera línea para eliminar de forma efectiva los inhibidores es prednisolona asociada a ciclofosfamida, con el que se describe una adecuada respuesta en 60-70% de los pacientes35-40. Como segunda línea de tratamiento se indica el uso de fármacos biológicos como rituximab, y como alternativa se recomienda la ciclosporina, micofenolato mofetilo, azatioprina y vincristina35-40.
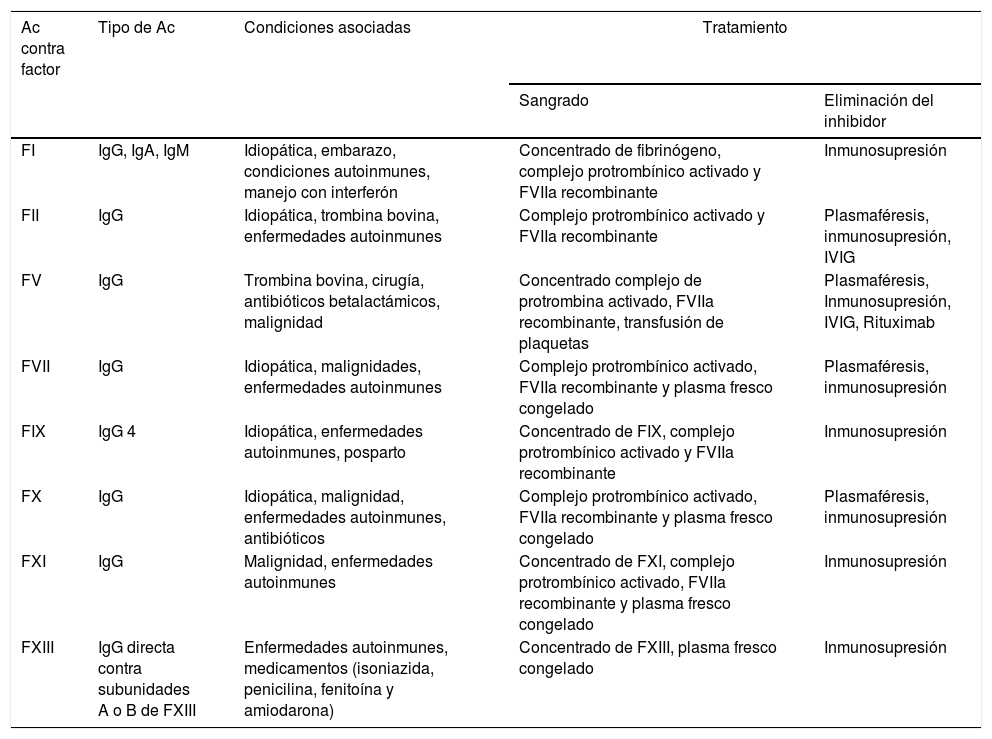
Como se mencionó previamente, también se han descrito inhibidores adquiridos dirigidos contra otros factores de la coagulación (I, II, V, VII, IX, X, XI y XIII)38. La severidad de las manifestaciones hemorrágicas se relaciona con el factor comprometido y, así mismo, el tratamiento está dirigido a lograr un adecuado control del sangrado y la eliminación del inhibidor40-43.
El síndrome de hipoprotrombinemia asociada anticoagulante lúpico se caracteriza por la presencia de anticuerpos no neutralizantes dirigidos contra el FII39-42. Se presenta con mayor proporción en el género femenino y suele manifestarse con hemorragias severas que incluyen la afección del sistema nervioso central. La sospecha clínica se da ante presencia de una prolongación significativa PT y PTTa, con una mínima corrección en la prueba de mezclas y la evidencia de anticoagulante lúpico40,41. Aunque no existe un manejo estandarizado, se recomienda la administración de plasma fresco o concentrados del complejo de protrombina asociado a tratamiento inmunosupresor40,42,43.
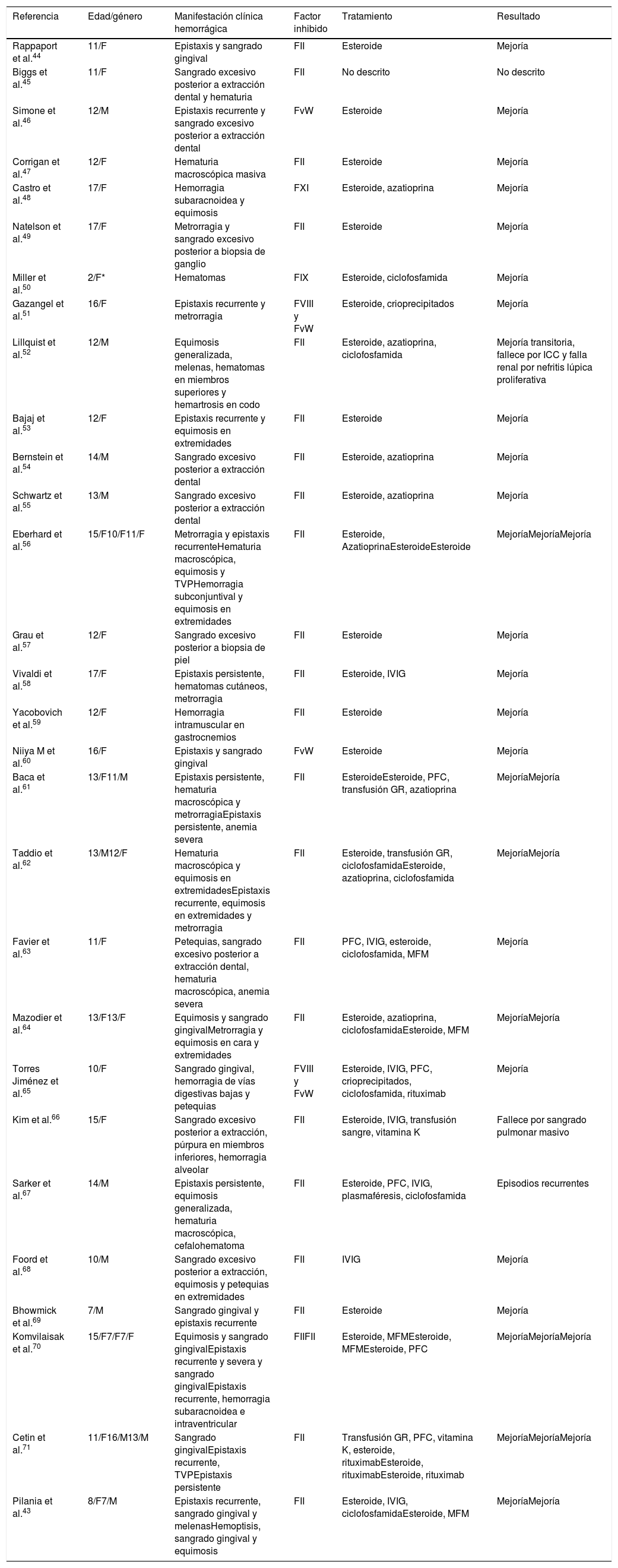
En la literatura se han publicado reportes de casos relacionados con la presencia de inhibidores y LESj. En la tabla 2 se describen las manifestaciones hemorrágicas presentes en pacientes con inhibidores asociados a LESj, el factor de coagulación comprometido y el requerimiento farmacológico de los pacientes reportados en la literatura y en la tabla 3 se presentan las características de los inhibidores para los otros factores y las enfermedades asociadas. Adicionalmente, se describe el tratamiento farmacológico recomendado tanto para control de sangrado como para erradicación o eliminación del inhibidor40.
Revisión de casos de la literatura relacionados con la presencia de inhibidores dirigidos contra los factores de coagulación en LESj
| Referencia | Edad/género | Manifestación clínica hemorrágica | Factor inhibido | Tratamiento | Resultado |
|---|---|---|---|---|---|
| Rappaport et al.44 | 11/F | Epistaxis y sangrado gingival | FII | Esteroide | Mejoría |
| Biggs et al.45 | 11/F | Sangrado excesivo posterior a extracción dental y hematuria | FII | No descrito | No descrito |
| Simone et al.46 | 12/M | Epistaxis recurrente y sangrado excesivo posterior a extracción dental | FvW | Esteroide | Mejoría |
| Corrigan et al.47 | 12/F | Hematuria macroscópica masiva | FII | Esteroide | Mejoría |
| Castro et al.48 | 17/F | Hemorragia subaracnoidea y equimosis | FXI | Esteroide, azatioprina | Mejoría |
| Natelson et al.49 | 17/F | Metrorragia y sangrado excesivo posterior a biopsia de ganglio | FII | Esteroide | Mejoría |
| Miller et al.50 | 2/F* | Hematomas | FIX | Esteroide, ciclofosfamida | Mejoría |
| Gazangel et al.51 | 16/F | Epistaxis recurrente y metrorragia | FVIII y FvW | Esteroide, crioprecipitados | Mejoría |
| Lillquist et al.52 | 12/M | Equimosis generalizada, melenas, hematomas en miembros superiores y hemartrosis en codo | FII | Esteroide, azatioprina, ciclofosfamida | Mejoría transitoria, fallece por ICC y falla renal por nefritis lúpica proliferativa |
| Bajaj et al.53 | 12/F | Epistaxis recurrente y equimosis en extremidades | FII | Esteroide | Mejoría |
| Bernstein et al.54 | 14/M | Sangrado excesivo posterior a extracción dental | FII | Esteroide, azatioprina | Mejoría |
| Schwartz et al.55 | 13/M | Sangrado excesivo posterior a extracción dental | FII | Esteroide, azatioprina | Mejoría |
| Eberhard et al.56 | 15/F10/F11/F | Metrorragia y epistaxis recurrenteHematuria macroscópica, equimosis y TVPHemorragia subconjuntival y equimosis en extremidades | FII | Esteroide, AzatioprinaEsteroideEsteroide | MejoríaMejoríaMejoría |
| Grau et al.57 | 12/F | Sangrado excesivo posterior a biopsia de piel | FII | Esteroide | Mejoría |
| Vivaldi et al.58 | 17/F | Epistaxis persistente, hematomas cutáneos, metrorragia | FII | Esteroide, IVIG | Mejoría |
| Yacobovich et al.59 | 12/F | Hemorragia intramuscular en gastrocnemios | FII | Esteroide | Mejoría |
| Niiya M et al.60 | 16/F | Epistaxis y sangrado gingival | FvW | Esteroide | Mejoría |
| Baca et al.61 | 13/F11/M | Epistaxis persistente, hematuria macroscópica y metrorragiaEpistaxis persistente, anemia severa | FII | EsteroideEsteroide, PFC, transfusión GR, azatioprina | MejoríaMejoría |
| Taddio et al.62 | 13/M12/F | Hematuria macroscópica y equimosis en extremidadesEpistaxis recurrente, equimosis en extremidades y metrorragia | FII | Esteroide, transfusión GR, ciclofosfamidaEsteroide, azatioprina, ciclofosfamida | MejoríaMejoría |
| Favier et al.63 | 11/F | Petequias, sangrado excesivo posterior a extracción dental, hematuria macroscópica, anemia severa | FII | PFC, IVIG, esteroide, ciclofosfamida, MFM | Mejoría |
| Mazodier et al.64 | 13/F13/F | Equimosis y sangrado gingivalMetrorragia y equimosis en cara y extremidades | FII | Esteroide, azatioprina, ciclofosfamidaEsteroide, MFM | MejoríaMejoría |
| Torres Jiménez et al.65 | 10/F | Sangrado gingival, hemorragia de vías digestivas bajas y petequias | FVIII y FvW | Esteroide, IVIG, PFC, crioprecipitados, ciclofosfamida, rituximab | Mejoría |
| Kim et al.66 | 15/F | Sangrado excesivo posterior a extracción, púrpura en miembros inferiores, hemorragia alveolar | FII | Esteroide, IVIG, transfusión sangre, vitamina K | Fallece por sangrado pulmonar masivo |
| Sarker et al.67 | 14/M | Epistaxis persistente, equimosis generalizada, hematuria macroscópica, cefalohematoma | FII | Esteroide, PFC, IVIG, plasmaféresis, ciclofosfamida | Episodios recurrentes |
| Foord et al.68 | 10/M | Sangrado excesivo posterior a extracción, equimosis y petequias en extremidades | FII | IVIG | Mejoría |
| Bhowmick et al.69 | 7/M | Sangrado gingival y epistaxis recurrente | FII | Esteroide | Mejoría |
| Komvilaisak et al.70 | 15/F7/F7/F | Equimosis y sangrado gingivalEpistaxis recurrente y severa y sangrado gingivalEpistaxis recurrente, hemorragia subaracnoidea e intraventricular | FIIFII | Esteroide, MFMEsteroide, MFMEsteroide, PFC | MejoríaMejoríaMejoría |
| Cetin et al.71 | 11/F16/M13/M | Sangrado gingivalEpistaxis recurrente, TVPEpistaxis persistente | FII | Transfusión GR, PFC, vitamina K, esteroide, rituximabEsteroide, rituximabEsteroide, rituximab | MejoríaMejoríaMejoría |
| Pilania et al.43 | 8/F7/M | Epistaxis recurrente, sangrado gingival y melenasHemoptisis, sangrado gingival y equimosis | FII | Esteroide, IVIG, ciclofosfamidaEsteroide, MFM | MejoríaMejoría |
Paciente con diagnóstico de enfermedad autoinmune con ANA positivos, alta sospecha LESj.
F: género femenino; FvW: factor von Willebrand; GR: glóbulos rojos; ICC: insuficiencia cardiaca congestiva; IVIG: inmunoglobulina intravenosa; M: género masculino; MFM: micofenolato mofetilo; PFC: plasma fresco congelado; TVP: trombosis venosa profunda.
Características de inhibidores adquiridos de los factores de coagulación
| Ac contra factor | Tipo de Ac | Condiciones asociadas | Tratamiento | |
|---|---|---|---|---|
| Sangrado | Eliminación del inhibidor | |||
| FI | IgG, IgA, IgM | Idiopática, embarazo, condiciones autoinmunes, manejo con interferón | Concentrado de fibrinógeno, complejo protrombínico activado y FVIIa recombinante | Inmunosupresión |
| FII | IgG | Idiopática, trombina bovina, enfermedades autoinmunes | Complejo protrombínico activado y FVIIa recombinante | Plasmaféresis, inmunosupresión, IVIG |
| FV | IgG | Trombina bovina, cirugía, antibióticos betalactámicos, malignidad | Concentrado complejo de protrombina activado, FVIIa recombinante, transfusión de plaquetas | Plasmaféresis, Inmunosupresión, IVIG, Rituximab |
| FVII | IgG | Idiopática, malignidades, enfermedades autoinmunes | Complejo protrombínico activado, FVIIa recombinante y plasma fresco congelado | Plasmaféresis, inmunosupresión |
| FIX | IgG 4 | Idiopática, enfermedades autoinmunes, posparto | Concentrado de FIX, complejo protrombínico activado y FVIIa recombinante | Inmunosupresión |
| FX | IgG | Idiopática, malignidad, enfermedades autoinmunes, antibióticos | Complejo protrombínico activado, FVIIa recombinante y plasma fresco congelado | Plasmaféresis, inmunosupresión |
| FXI | IgG | Malignidad, enfermedades autoinmunes | Concentrado de FXI, complejo protrombínico activado, FVIIa recombinante y plasma fresco congelado | Inmunosupresión |
| FXIII | IgG directa contra subunidades A o B de FXIII | Enfermedades autoinmunes, medicamentos (isoniazida, penicilina, fenitoína y amiodarona) | Concentrado de FXIII, plasma fresco congelado | Inmunosupresión |
Ac: anticuerpos; Ig: inmunoglobulina; IVIG: inmunoglobulina intravenosa.
Fuente: Tomado de Franchinni et al.40.
Las alteraciones hematológicas son comunes en los pacientes con LES. Es importante diferenciar entre las manifestaciones relacionadas con la enfermedad y aquellas que son secundarias al tratamiento. El compromiso hematológico puede diferenciarse según si se relaciona con el compromiso de las líneas celulares, evidenciado con la presencia de leucocitopenia, trombocitopenia, anemia (anemia hemolítica, anemia de enfermedad crónica y anemia por deficiencia de hierro) y alteraciones relacionadas con el compromiso de la coagulación.
El compromiso en la coagulación se asocia con manifestaciones trombóticas y hemorrágicas. La presencia de aPL favorece la presencia de manifestaciones trombóticas, a nivel arterial o venoso, y manifestaciones no trombóticas frecuentemente descritas en pacientes con SAF aislado o asociado a LES. Las manifestaciones no trombóticas incluyen hallazgos hematológicos (trombocitopenia inmune, anemia hemolítica con prueba de Coombs positiva y leucocitopenia), compromiso cutáneo (fenómeno de Raynaud, livedo reticularis, úlceras cutáneas) y compromiso neurológico (cefalea migrañosa, desórdenes del movimiento, epilepsia y desórdenes del afecto en menor frecuencia). Existen factores de riesgo adicionales a la presencia de aPL que favorecen el desarrollo de manifestaciones trombóticas como la presencia de niveles elevados de homocisteína (secundaria de déficit de cianocobalamina, falla renal crónica, hipotiroidismo y mutaciones en el gen MTHFR), déficit adquirido de la proteína S, proteína C y antitrombina.
La diátesis hemorrágica también se ha descrito en menor frecuencia. La hemorragia alveolar es una manifestación rara, pero con alta mortalidad, que requiere un diagnóstico precoz y tratamiento agresivo. Además, las manifestaciones hemorrágicas también se relacionan con el déficit adquirido de los factores de la coagulación en relación con la presencia de inhibidores. Se estima que aproximadamente el 16% de los casos de inhibidores adquiridos se presentan en el contexto de enfermedades autoinmunes, con una mayor proporción en pacientes con LES. Es importante tener un alto índice de sospecha ante pacientes con trastornos hemorrágicos, sin antecedente familiar o personal de hemorragias, con tiempos de coagulación prolongados que no corrigen con la prueba de mezclas.
El diagnóstico precoz y tratamiento temprano disminuyen la morbimortalidad relacionada con este tipo de manifestaciones en LES.
Conflicto de interesesNinguno de los autores declara conflictos de interés.
