



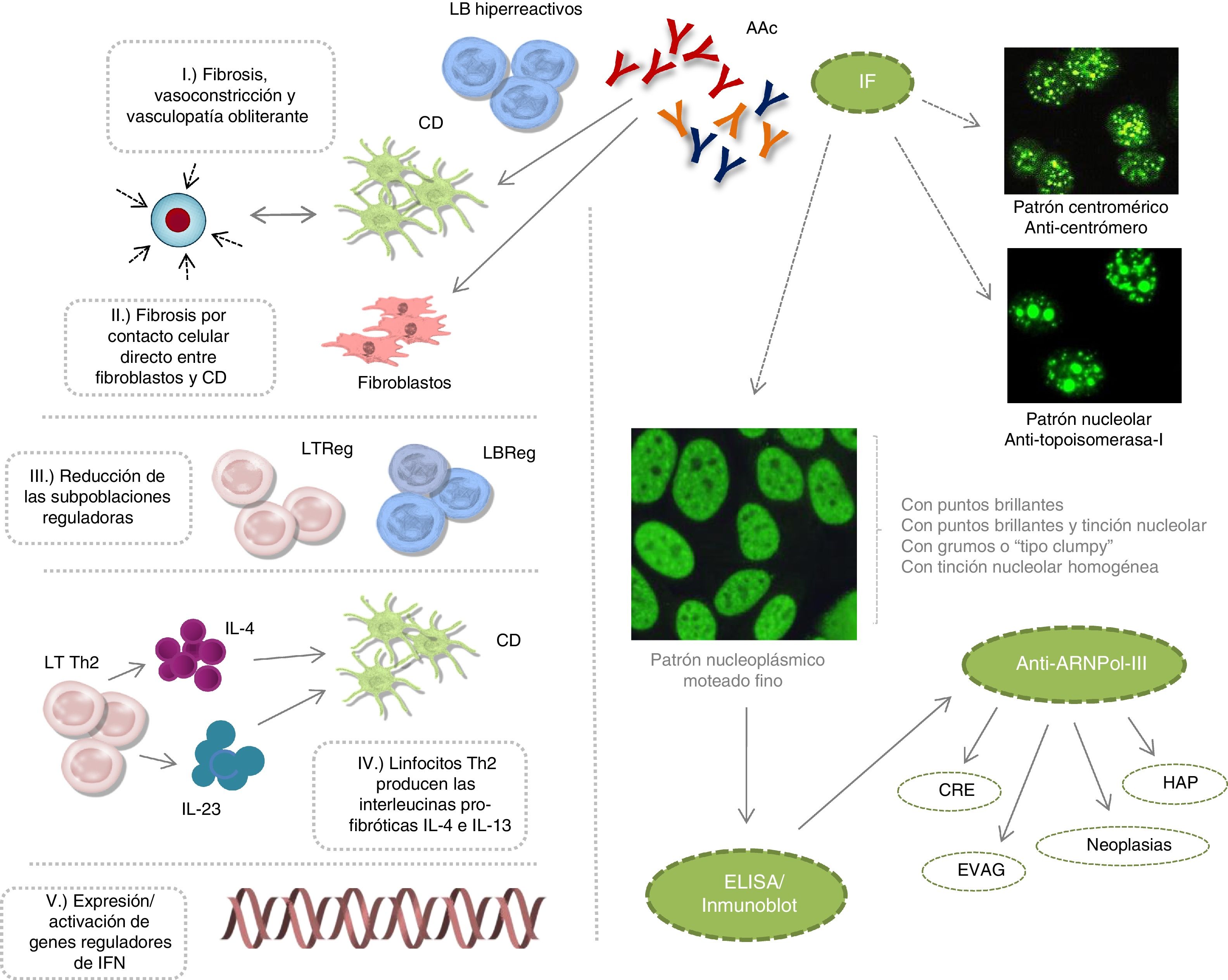
La esclerosis sistémica cutánea (ESC) es un trastorno crónico del tejido conectivo de origen autoinmune, caracterizado por lesiones microvasculares, depósito de colágeno y fibrosis de la piel y las vísceras incluyendo: tracto digestivo, pulmones, corazón, riñones. La detección de autoanticuerpos (AAc) circulantes dirigidos contra antígenos nucleares (ANA) es importante en el diagnóstico de la enfermedad y de sus formas clínicas, ESC difusa y ESC localizada.
A pesar de que estos AAc participan en la fisiopatología de la ESC, no se han descrito los mecanismos específicos que favorecen la desregulación del sistema inmunológico. Actualmente, se postula que: 1) la fibrosis es promovida por linfocitos B hiperreactivos productores de autoanticuerpos, la maduración de los fibroblastos o la inhibición de la degradación de la matriz extracelular, junto a eventos de vasoconstricción y vasculopatía obliterante; 2) los linfocitos B promueven la fibrosis por medio del contacto celular directo con fibroblastos y células dendríticas; 3) la reducción de linfocitos B y T con perfil regulador favorecen la formación de fibrosis tisular; 4) linfocitos T de tipo Th2 producen interleucinas profibróticas IL-4 e IL-13 que inducen la maduración de células dendríticas y retroalimentan este microambiente profibrótico y 5) un incremento en la expresión/activación de genes reguladores del interferón inducen la fibrosis tisular1–3 (fig. 1).

Eventos etiopatogénicos de la ESC, patrones de IF y producción de los autoanticuerpos: anticentrómero, antitopoisomerasa-I y anti-ARNPol-III.
AAc: autoanticuerpos; CD: célula dendrítica; CRE: crisis renal esclerodérmica; EVAG: ectasia vascular antral gástrica; HAP: hipertensión arterial pulmonar; IF: inmunofluorescencia; IFN: interferón; IL: interleucina; LB: linfocito B; LT: linfocito T; Reg: regulador.
Los eventos clínicos (fenómeno de Raynaud), histológicos (microvasculopatía), celulares (apoptosis de las células endoteliales) o autoinmunes (desarrollo de AAc) pueden aparecer años antes de la fibrosis cutánea1,2. Entre 80-98% de los pacientes con AAc, presentan en la prueba de ANA alguno de estos patrones de inmunofluorescencia: centromérico, nucleolar o nuclear moteado fino. Se ha descrito que estos AAc van dirigidos específicamente contra proteínas centroméricas (anticentrómero), enzimas como la topoisomerasa-I (anti-Scl-70) o enzimas encargadas de transcribir el ADN en ARN, como la enzima polimerasa nucleotidiltransferasa del ácido ribonucleico III (anti-ARNPol-III). Otros AAc que pueden aparecer, aunque con menor frecuencia, son los denominados antirribonucleoproteínas Th/To, anti-PM-Scl y antifibrilarina (U3-RNP)4–7.
La importancia de estas especificidades se debe a que la frecuencia de cada uno de estos AAc varía en función del grupo étnico y de la procedencia geográfica de los pacientes, también han sido asociados con distintos perfiles clínicos y no suelen coexistir en el mismo paciente ni cambiar durante el curso de la enfermedad. Motivos estos por los cuales su determinación en el momento del diagnóstico resulta de gran utilidad para predecir tanto la afectación orgánica como la evolución clínica de nuestros pacientes.
Los antitopoisomerasa-I y anticentrómero se asocian con ESC difusa y ESC localizada, respectivamente. Los anticuerpos anti-Th/To, anti-PM-Scl y antifibrilarina se han detectado en pacientes con 1) fibrosis e hipertensión pulmonar o polimiositis, 2) lupus eritematoso sistémico y 3) síndrome de solapamiento, respectivamente6,8. Lazzaroni et al. en un estudio promovido por European Scleroderma Trials and Research han demostrado que la crisis renal esclerodérmica, la ectasia vascular antral gástrica, la progresión rápida del espesor de la piel, la hipertensión arterial pulmonar y las neoplasias son características clínicas independientes asociadas con la presencia de anti-ARNPol-III9. La importancia de estas asociaciones ha promovido la inclusión de los anti-ARNPol-III en los criterios más recientes de clasificación de la ESC elaborados por un consenso de expertos del American College of Rheumatology y la European League against Rheumatism10. Por lo tanto, en la práctica clínica habitual además de la determinación de AAc antitopoisomerasa-I y anticentrómero, sería de gran utilidad y más que recomendable disponer siempre del ensayo para la detección de anti-ARNPol-III. Igualmente, sería de importancia cardinal su inclusión en el diseño de los ensayos clínicos y en los protocolos de seguimiento de nuestros pacientes.
