



Las miopatías inflamatorias idiopáticas (MII) constituyen un grupo heterogéneo de enfermedades de origen autoinmune caracterizadas por afectar a la musculatura estriada y por su naturaleza inflamatoria1,2. Se incluyen en este grupo la dermatomiositis, la polimiositis y la miositis con cuerpos de inclusión. Aunque el músculo es el principal órgano afecto, el pulmón, entre otros órganos internos, también se afecta con frecuencia, siendo la enfermedad pulmonar intersticial (EPI) la manifestación respiratoria más frecuente1,2. La presencia de hipertensión arterial pulmonar (HAP) es infrecuente3,4. Hasta la fecha, solo existen casos aislados publicados en la literatura de pacientes con hipertensión pulmonar (HP) asociada a MII y prácticamente todos corresponden a pacientes con EPI grave, siendo la HP secundaria (HP de grupo3)5-7.
Presentamos el caso de una paciente de 48años, sin factores de riesgo cardiovascular, con historia de asma bronquial persistente leve, bien controlada con tratamiento inhalado y diagnosticada de dermatomiositis-polimiositis a los 36años, en seguimiento por reumatología, sin signos de actividad actual ni tratamiento activo. Refería historia de alteraciones vasculares cutáneas desde la infancia, así como de reflujo gastroesofágico e hipotiroidismo. No presentaba antecedentes epidemiológicos de interés y seguía tratamiento habitual con fluticasona/vilanterol, omeprazol, levotiroxina, celecoxib y metamizol.
La paciente ingresó para estudio de disnea progresiva de meses de evolución hasta hacerse de mínimos esfuerzos, asociada a dolor opresivo en hemitórax izquierdo y palpitaciones con los esfuerzos. No refería ortopnea ni disnea paroxística nocturna, ni presentaba datos de congestión periférica. A la exploración permanecía taquipneica al habla y toleraba el decúbito. La presión arterial era de 109/75mmHg, la frecuencia cardiaca de 92lpm y la saturación basal de oxígeno del 90%. En la auscultación cardiorrespiratoria presentaba un soplo sistólico sugerente de insuficiencia tricuspídea (IT) y aumento de la intensidad del segundo tono. El abdomen era normal y no tenía edemas. Como pruebas complementarias iniciales se solicitó una analítica completa con NT-proBNP 3010 pg/l, con resto de bioquímica, hemograma y coagulación normal. Las serologías para virus hepatotropos y virus de la inmunodeficiencia humana (VIH) fueron negativas, así como el estudio de autoinmunidad. La gasometría basal mostraba una hipoxemia moderada (pO2: 63mmHg) sin otras alteraciones. El ecocardiograma objetivaba un ventrículo izquierdo (VI) de morfología y función normales. El ventrículo derecho (VD) estaba dilatado (43mm) y mostraba signos de disfunción sistólica (TAPSE 13mm, s’ 8cm/s, FAC 25%). La aurícula derecha (AD) tenía un área de 22cm2. Existía una IT ligera que permitía estimar una presión sistólica de la arteria pulmonar (PsAP) de 85mmHg. Se observaba un derrame pericárdico ligero sin signos de compromiso hemodinámico. No se evidenciaron shunts. La vena cava inferior no estaba dilatada y mostraba un colapso inspiratorio adecuado.
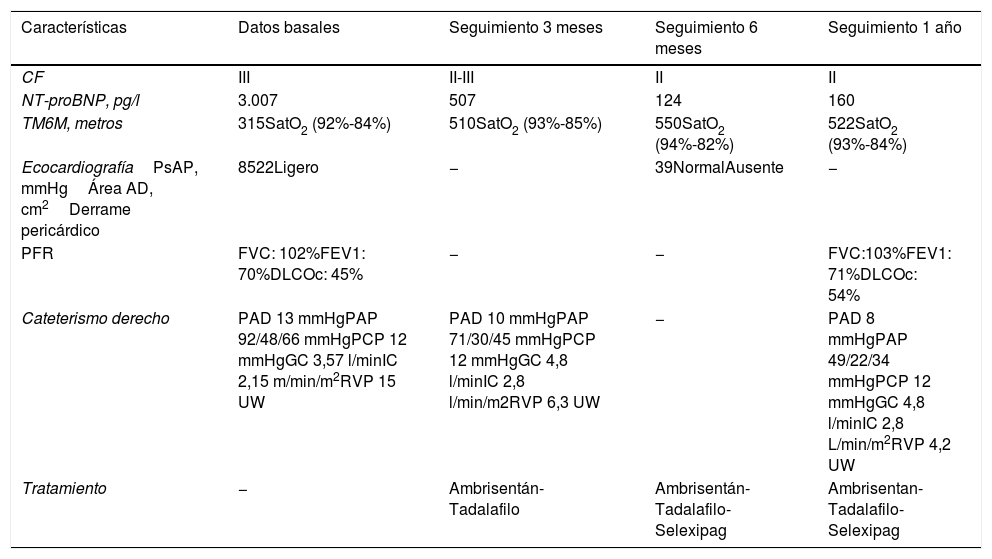
Con el diagnóstico de HP con afectación severa del VD e hipoxemia secundaria se inició el estudio etiológico con ecografía abdominal normal, pruebas de función respiratoria (PFR) con los siguientes resultados: capacidad vital forzada (FCV):102%, volumen espiratorio máximo en el primer segundo de la espiración (FEV1): 70%, cociente FEV1/FVC: 68, capacidad pulmonar total (TLC): 101%, capacidad de difusión de monóxido de carbono (DLCOc): 45%. Además se realizó una gammagrafía de perfusión que fue normal y una TAC torácica sin signos de enfermedad intersticial ni enfermedad venooclusiva pulmonar (EVOP). Por último se practicó un cateterismo derecho (CD) que confirmó el diagnostico de HP grave con los siguientes parámetros: presión de aurícula derecha (PAD) 13mmHg, presión de arteria pulmonar (PAP) 92/48/66mmHg, presión de enclavamiento pulmonar (PCP) 12mmHg, gasto cardiaco (GC) 3,57l/min, índice cardiaco (IC) 2,15l/min/m2, resistencias vasculares pulmonares (RVP) 15unidades Wood (UW). La capacidad funcional se evaluó con el test de marcha de 6min (TM6M), en el que la paciente caminó 315m y se detuvo a los 2min por disnea y desaturación al 80%. El electromiograma de control mostró un patrón miopático generalizado crónico sin signos de actividad. Con los resultados de las exploraciones complementarias se estableció el diagnostico de HAP asociada a PM-DM con perfil de riesgo intermedio. Se inició tratamiento con oxigenoterapia, diuréticos y vasodilatador específico con ambrisentán y tadalafilo, con buena tolerancia clínica y analítica. La tabla 1 muestra los resultados de las exploraciones complementarias durante el seguimiento. A los 3meses la paciente presentaba mejoría clínica y funcional con una clase funcional (CF) II-III, sin episodios de síncope ni dolor torácico ni signos de congestión sistémica. A pesar de la mejoría clínica, dada la persistencia en situación de riesgo intermedio se añadió al tratamiento selexipag. Al año de seguimiento la paciente se mantenía estable desde el punto de vista cardiorrespiratorio y de capacidad de esfuerzo con CF II/IV, sin síncopes ni dolor centrotorácico ni sintomatología congestiva con el tratamiento diurético pautado. Las pruebas de control revelaron mejoría funcional y hemodinámica.
Características clínicas, funcionales y hemodinámicas al diagnóstico y durante el seguimiento
| Características | Datos basales | Seguimiento 3 meses | Seguimiento 6 meses | Seguimiento 1 año |
|---|---|---|---|---|
| CF | III | II-III | II | II |
| NT-proBNP, pg/l | 3.007 | 507 | 124 | 160 |
| TM6M, metros | 315SatO2 (92%-84%) | 510SatO2 (93%-85%) | 550SatO2 (94%-82%) | 522SatO2 (93%-84%) |
| EcocardiografíaPsAP, mmHgÁrea AD, cm2Derrame pericárdico | 8522Ligero | − | 39NormalAusente | − |
| PFR | FVC: 102%FEV1: 70%DLCOc: 45% | − | − | FVC:103%FEV1: 71%DLCOc: 54% |
| Cateterismo derecho | PAD 13 mmHgPAP 92/48/66 mmHgPCP 12 mmHgGC 3,57 l/minIC 2,15 m/min/m2RVP 15 UW | PAD 10 mmHgPAP 71/30/45 mmHgPCP 12 mmHgGC 4,8 l/minIC 2,8 l/min/m2RVP 6,3 UW | − | PAD 8 mmHgPAP 49/22/34 mmHgPCP 12 mmHgGC 4,8 l/minIC 2,8 L/min/m2RVP 4,2 UW |
| Tratamiento | − | Ambrisentán-Tadalafilo | Ambrisentán-Tadalafilo-Selexipag | Ambrisentan-Tadalafilo- Selexipag |
AD: aurícula derecha; CF: clase funcional; DLCOc: capacidad de difusión de monóxido de carbono; FEV1: volumen espiratorio máximo en el primer segundo de la espiración; FVC: capacidad vital forzada; GC: gasto cardiaco; IC: índice cardiaco; PAD: presión aurícula derecha, PAP: presión arteria pulmonar; PCP: presión de enclavamiento pulmonar; PFR: pruebas de función respiratoria; PsAP: presión sistólica arteria pulmonar; RVP: resistencias vasculares pulmonares; SatO2: saturación de oxígeno; TM6M: test de marcha 6min; UW: unidades Wood.
La hipertensión arterial pulmonar (HAP) es una complicación infrecuente y tardía de las MII8-13. Su patogenia no está del todo aclarada, aunque se especula con la afectación microvascular del lecho pulmonar por el proceso inflamatorio difuso de la MII1,14. Se debe sospechar en caso de disnea, síncope o fallo derecho, especialmente en pacientes con afectación cutánea, microangiopatía periférica y anticuerpos anti-SSA positivos. Si bien existen casos aislados descritos en la literatura, siempre hay que descartar otras causas antes de atribuir la HP a una afectación vascular primaria en estos pacientes8. El abordaje terapéutico no está bien definido. No obstante, el uso conjunto de inmunosupresores y vasodilatadores específicos en los enfermos con miopatía activa e HAP se ha asociado a un mejor control de la enfermedad8,15. La estratificación pronóstica de la HAP y la estrategia de tratamiento orientado por objetivos, tal y como recomiendan las guías de práctica clínica, son útiles y permiten adelantarnos al deterioro clínico y conseguir un mayor control de la enfermedad.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses en relación con este escrito.
