



Introduction
Isolated 3-Methylcrotonyl-CoA Carboxylase deficiency (3-MCC) is an inborn error of leucine catabolism inherited in an autosomal recessive pattern.1 In 1997 only 26 cases had been reported.2 Since the introduction of MS/MS to newborn screening programs the diagnosis of 3-MCC deficiency has increased. Patients with this disorder show a highly variable clinical phenotype, ranging from asymptomatic detected when the family is tested because a newborn is diagnosed with 3-MCC deficiency to a severe clinical presentation that includes recurrent episodes of vomiting, lethargy and hypotonia. If untreated, this disorder can lead to developmental delay, seizures, coma and death. Early detection and lifelong management may prevent many of the complications that are often triggered by increased ingestion of proteins or infection. The metabolic stress to which these individuals are exposed is an important factor in the onset of the disease and the variability of symptoms. The diagnosis is made by a characteristic biochemical profile that includes plasma elevation of 3-hydroxyisovaleryl-carnitine (C5OH) and low levels of carnitine detected by MS/MS, as well as increased urine levels of 3-methylcrotonyl-glycine (3 MCG) and 3-hydroxyisovaleric acid (3-HIVA) detected by gas chromatography-mass spectrometry (GC/MS). The confirmatory test is based on the detection of decreased enzyme activity in white cells or fibroblasts. The analyte C5OH is also positive in other leucine and isoleucine disorders such as 3-Methylglutaconic hydratase (MGA), Hydroxy-3-methylglutaryl-CoA lyase (HMG), 2-Methyl-3-hydroxybutyryl- CoA Dehydrogenase, beta-ketothiolase deficiency, multiple carboxylase deficiency and that due to maternal transfer or liver immaturity in the newborn.
The introduction of tandem mass spectrometry (MS/ MS) to expand the neonatal screening program in Nuevo Leon, a state in northeastern Mexico, has eliminated several of the problems associated with previous screening technology such as high false positive rates, the high cost of individual testing and the performance of a single test to diagnose each disease was time consuming.3 This has permitted the early detection of a greater number of amino acid, organic acid and fatty acid oxidation disorders.
In 2003 Koeberl et al4 reported 8 cases of 3-MCC in North Carolina with a higher incidence among Hispanics. This encouraged us to study the incidence of 3-MCC deficiency in our population. In this paper we report the findings of abnormal results of C5OH detected by MS/MS associated with isolated 3-Methylcrotonyl-CoA Carboxylase deficiency (3-MCC).
Materials and methods
From 2002 to 2004, all the hospitals belonging to the Secretariat of Health of the state of Nuevo Leon started a pilot study using MS/MS in the newborn screening program. There are around 90,000 births per year in Nuevo Leon and one fourth of them are born in hospitals belonging to the State Secretariat of Health.
Samples: Blood samples were obtained by heel prick collected during the first 24 hours of life spotted on filter paper cards (S&S 903) and allowed to dry. All cards had the following data: mother's name, newborn´s DOB, hospital of birth, gestational age, sex, weight, height, breast or bottle feeding, date and hour. Samples were sent to the Genetics Department of the Medical School, Autonomous University of Nuevo Leon. Samples were extracted and processed according to standard methods and if one flagged for abnormal elevation of 3-hydroxyisovalerylcarnitine (C5OH) the newborn was retested at 2 to 3 weeks of age. If C5OH levels were again elevated, follow-up tests such as a plasma acylcarnitine profile and urine organic acids were obtained according to the standard algorithm for the diagnosis of isolated 3-MCC deficiency.5 Family members were also tested.
Preparation of sample: a blood specimen is punched into a 1/8-inch disk (3 uL of blood on filter paper). Each disk is placed into wells of a 96-well microtiter plate. The disks are mixed in methanol containing 12 stable isotope internal standards of acylcarnitines and 12 stable isotope internal standards of amino acids (NeoGram Amino Acids and Acylcarnitines Tandem Mass Spectrometry Kit). The plates are incubated at 30o C and shaken at 750 rpm for 30 minutes. A solution (50 uL) of hydrochloric acid in anhydrous n-butanol is added to each well, mixed and sealed; the metabolites with deuterated internal standards are converted into the corresponding n-butyl esters by heating at 65 oC for 30 minutes.
MS/MS: Amino acids and acylcarnitines (AC) are analyzed as butyl esters by using MS/MS triple-quadropole API 2000 (PerkinElmer Sciex) that is connected with a pump and autosampler 200/ PerkinElmer. Briefly, the analysis is carried out on small aliquots (3 uL) of each reconstituted specimen that are injected into an ion chamber; each AC is calculated on the basis of its stable-isotope deuterium compound.
All data were obtained and processed utilizing Analyst software and the NeoGram database. The mass spectrum is converted to a significant clinical result by calculating the concentration of each analyte. The results are expressed in uM/L.
The normal values for our population were previously obtained, the normal value of C5OH was established at 0.054-1.2 uM/L and all patients with values >3 SD were considered abnormal and another sample was requested in order to confirm the presumptive abnormality.6
GC/MC analysis: Urine organic acid determination was performed in all newborns with isolated elevations of C5OH in the newborn screening. Increased levels of urine 3 methylcrotonylglycine (3 MCG) and 3-hydroxyisovaleric acid (3-HIV) established the diagnosis of 3-MCC deficiency. GC/MC analysis was performed in accordance with the standard technique that is well-documented by Honour.7
Results
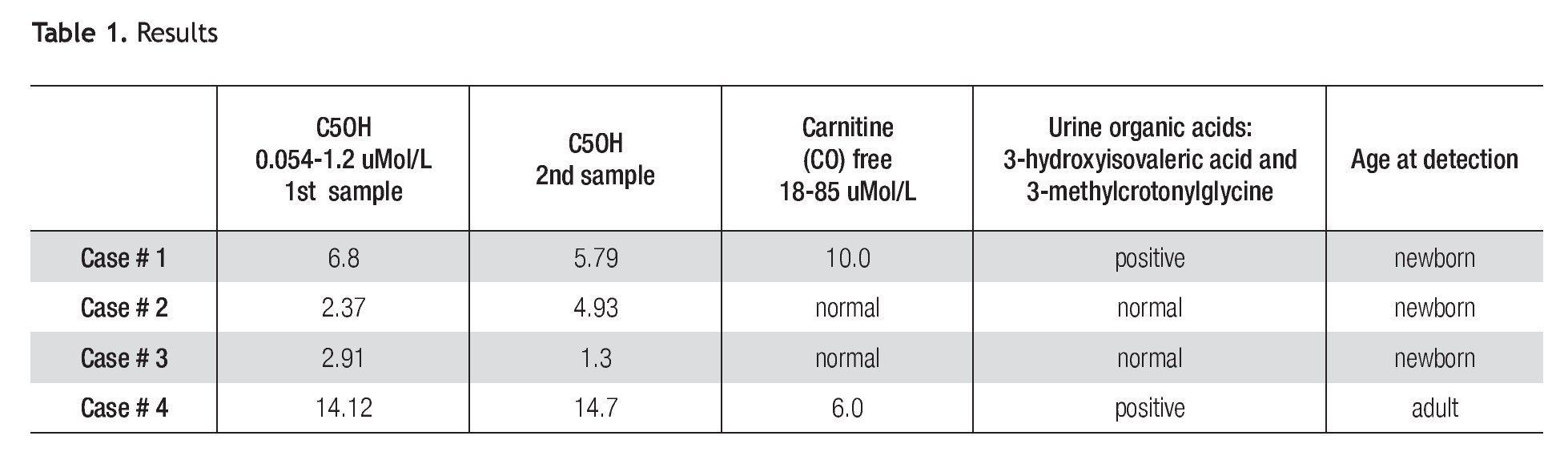
A total of 42,264 newborns were screened. We found 3 cases of elevation of C5OH (Table 1). All individuals were contacted and referred to the Genetics Department for a complete medical evaluation.
Clinical description
Patient 1 is a full-term female neonate of non-consanguineous parents, normal delivery, weight of 3.730 Kg, height of 52 cm. The mother had one previous miscarriage and 4 normal children. The neonatal physical exam was normal and the baby was discharged from the hospital at 24 hours. An elevation of C5OH of 6.8 μMol/L (5x normal value) and low free carnitine was reported (Table 1). The second sample taken at 21 days of life was also abnormal (4.8 x normal value). The baby was referred to the Genetics Department. The urine organic acids (GC/ MS) reported high levels of 3 methylcrotonylglycine, and 3-hydroxyisovaleric acid (Table 1). The mother and all the family were negative in the MS/MS test.
Patient 2 is a full-term male baby of non-consanguineous parents, negative family history, normal delivery, weight of 3.050 Kg, height 50 cm, physical exam was normal and he was discharged at 24 hours; the C5OH level was abnormal at 2.37 μMol/L (2X). A second sample taken at 21 days was also reported at 4.93 μMol/L (4X). Urine organic acids and free carnitine blood levels were within normal values. Despite normal results of urine organic acids, C5OH persisted elevated until 5 months of age. After that, blood levels of C5OH normalized. His family was tested in order to rule out the possibility of asymptomatic 3-MCC deficiency, the results were normal.
Patient 3 is a full-term female baby, normal delivery, of non-consanguineous parents, family history negative, weight of 3.400 Kg, height of 52 cm, physical exam was normal and she was discharged at 24 hours. Levels of C5OH were 2.91 μMol/L (2.4 times elevated), free carnitine was normal. A second sample taken at 15 days showed a significant decrease of C5OH levels; blood free carnitine and urine organic acids were at normal levels. Her mother was tested and her results are shown as patient 4.
Patient 4 is a 35 year old female, whose C5OH values was doubled and free carnitine was below normal. Her first test was taken 4 weeks after her baby was born and the results were 14.12 μ Mol/L (11.8 times) elevated); free carnitine was low. A second test was taken at a one month interval showing almost the same values (C5OH = 14.7 μMol/L and low free carnitine). Results are shown in Table 1. She denied symptoms suggestive of 3MCC deficiency, such as fatigue, muscle cramps or weakness and also denied episodes of hypoglycemia or epilepsy. Her physical exam was normal except for grade 1 obesity. The urine organic acids showed abnormal levels of 3-hydroxyisovaleric acid and 3-methylcrotonylglycine which suggested isolated 3-methylcronotonyl-CoA carboxylase deficiency.
These patients were followed every 30 days for 18 months, focusing on cases 1 and 4. Patient 1 with a restricted protein diet of 2 gr. /Kg/day and carnitine PO 50 mg/Kg/day is developing and growing well. Her weight, height and head circumference are normal. She has had episodes of viral gastroenteritis and also a common cold and she did well without complications such as metabolic acidosis or dehydration. All the infants are currently healthy. Growing and developing normally. The adult patient is asymptomatic.
Discussion
The primary aim of a newborn screening program is the early detection and treatment of clinically important disorders in order to minimize morbidity and mortality in early childhood.
The incidence of 3-MCC deficiency depends on the ethnic group screened. Before the era of expanded newborn screening by MS/MS, Weisman3 in 1998 had reported 26 cases of 3-MCC deficiency corroborated clinically and by urine organic acids. Wilken8 in 1998 reported 3 cases of MCC deficiency detected by MS/MS in newborn screening in Australia, although in his experience, he has not seen a symptomatic patient with this disorder.
Koeberl5 et al 2003 reported an incidence of 1/64000, in North Carolina mainly in Hispanics. He detected eight newborns with 3-MCC deficiency; three of them (37.5%) were Hispanics. The New England Program reported an incidence of 1/60,0009 while Rashed after ten years of screening in Saudi Arabia found only one case.4 In our study the incidence of 3-MCC deficiency is higher than in Caucasians. In our report one baby had the disease while in the others elevation of C5OH was due to liver immaturity (Case 2) and maternal transfer (Case 3). Case 4 confirmed the importance of screening the family in order to provide genetic counseling.
It is of note that case 1 and 4, in which the disease was confirmed, abnormal levels of C5OH were higher than in the other two cases. Although it is possible that baby 1 may never have clinically significant disease, long-term follow up and treatment as described in symptomatic cases is necessary. The adult case in this report probably has a benign disease because despite the stress of giving birth, she did not present metabolic decompensation or any kind of symptom related with this disorder.
There are controversial issues regarding the need for treating these patients. Wilken (2003)8 excluded the report of positive cases of 3-MCC in Australia´s newborn screening program because he considered is a benign disease. Mourmans (1995),10 and Lehnert (1996)11 also reported it as a benign condition and Gibson (1998)12 reported positive asymptomatic cases of siblings and mothers of infants detected by newborn screening. Nevertheless, there are many other publications reporting severely ill patients with symptomatic 3-MCC deficiency.3,12-14
The outcome for 3-MCC deficiency is highly variable. Catastrophic acute presentations include severe hypoglycemia (Oude et al 2005)13 Reye-like symptoms or neurological non-acute symptoms such as psychomotor retardation, hypotonia, arreflexia, failure to thrive and leukodystrophy as described by Kremer (2002).14 Different presentations in a single family have been described by Visser15 in which he reported an infant with failure to thrive, gastro-esophageal reflux, hypertonia and dilated cardiomyopathy associated with carnitine deficiency, her brother had only developmental delays and their father was asymptomatic. These reflect the variable expressiveness of the disease.
Urinary organic acid analysis usually enables physicians to make the diagnosis, based on the finding of characteristic analytes. Confirmation of 3-MCC deficiency based on the determination of enzymatic activity in lymphocytes or fibroblasts is desirable; however, Koeberl,5 Roschinger16 and Van Hove17 have reported a wide range of results in the enzymatic activity that are not related to the symptoms and the organic acid levels. For the above mentioned reasons the determination of enzymatic activity could be unnecessary for the diagnosis. We concluded that the presence of C5OH in blood and of the organic acids 3-hydroxyisovaleric acid and 3-methylcrotonylglycine in urine would be enough to confirm the disease.
This is the first report of 3-MCC deficiency in Mexico. In order to know what would be the most common clinical phenotype in our population it is necessary to introduce the expanded newborn screening to other states of the country.
We conclude that the introduction of MS/MS technology in newborn screening programs will detect additional inborn errors of metabolism that otherwise remain un-diagnosed, such as 3-MCC deficiency, which is usually not tested in many newborn screening programs. This has enabled us to study this disease in our population in order to compare it with the literature. In very symptomatic patients tandem mass spectrometry is a rapid screening test that should be included early in the diagnostic work up of metabolic disorders.
Corresponding author: Dra. Enid Alda Rangel Córdova.
Departamento de Genética, Facultad de Medicina y Hospital Universitario Dr. José Eleuterio González de la UANL. Avenida Francisco I. Madero y Gonzalitos s/n, Colonia Mitras Centro. C. P. 64460. Monterrey, Nuevo León, México. Telephone: (+52 81) 8329 4217.
E-mail:enidcor2001@hotmail.com
Received: november, 2009. Accepted: november, 2009