














Despite their relatively low prevalence, vascular diseases of the liver represent a significant health problem in the field of liver disease. A common characteristic shared by many such diseases is their propensity to cause portal hypertension together with increased morbidity and mortality. These diseases are often diagnosed in young patients and their delayed diagnosis and/or inappropriate treatment can greatly reduce life expectancy.
This article reviews the current body of evidence concerning Budd–Chiari syndrome, non-cirrhotic portal vein thrombosis, idiopathic portal hypertension, sinusoidal obstruction syndrome, hepatic vascular malformations in hereditary haemorrhagic telangiectasia, cirrhotic portal vein thrombosis and other rarer vascular diseases including arterioportal fistulas. It also includes a section on the diagnostic imaging of vascular diseases of the liver and their treatment from a haematological standpoint (study of thrombotic diathesis and anticoagulation therapy). All recommendations are based on published studies extracted from PubMed. The quality of evidence and strength of recommendations were rated in accordance with the GRADE system (Grading of Recommendations, Assessment Development and Evaluation). In the absence of sufficient evidence, recommendations were based on the opinion of the committee that produced the guide.
Las enfermedades vasculares hepáticas, a pesar de su relativamente baja prevalencia, representan un problema de salud importante en el campo de las enfermedades hepáticas. Una característica común a muchas de estas enfermedades es que pueden causar hipertensión portal, con la elevada morbimortalidad que ello conlleva. Con frecuencia estas enfermedades se diagnostican en pacientes jóvenes y el retraso en su diagnóstico y/o un tratamiento inadecuado pueden reducir de forma importante la esperanza de vida.
El presente artículo revisa la evidencia actual en el síndrome de Budd-Chiari, la trombosis venosa portal en pacientes no cirróticos, la hipertensión portal idiopática, el síndrome de obstrucción sinusoidal, las malformaciones vasculares hepáticas en la telangiectasia hemorrágica hereditaria, la trombosis portal en la cirrosis, otras patologías vasculares menos frecuentes como las fístulas arterioportales, así como un apartado sobre el diagnóstico por imagen de las enfermedades vasculares hepáticas y su tratamiento desde el punto de vista hematológico (estudio de la diátesis trombótica y tratamiento anticoagulante). Las recomendaciones se han realizado de acuerdo a los estudios publicados extraídos de Pubmed. La calidad de la evidencia y la intensidad de las recomendaciones fueron graduadas de acuerdo al sistema Grading of Recommendations Assessment Development and Evaluation (GRADE). Cuando no existían evidencias suficientes, las recomendaciones se basaron en la opinión del comité que redactó la guía.
Despite their relatively low prevalence (less than 5/10,000 patients), vascular diseases of the liver represent a significant health problem in the field of liver disease. A common characteristic shared by many such diseases is that they can cause portal hypertension (PHT) along with the increased morbidity and mortality that this involves. These diseases are often diagnosed in young patients and delayed diagnosis and/or inappropriate treatment can greatly reduce life expectancy.
Advances in knowledge about these diseases are hampered by the small number of studies directed at evaluating their natural history, pathophysiology or treatment. Nevertheless, recently the interest in these diseases has increased as evidenced by the increase in the number of publications about the topic observed in recent years.
This article reviews the current body of evidence concerning Budd–Chiari syndrome, non-cirrhotic portal vein thrombosis, idiopathic PHT, sinusoidal obstruction syndrome, hepatic vascular malformations in hereditary haemorrhagic telangiectasia, cirrhotic portal vein thrombosis and other rarer vascular diseases including arterioportal fistulas. It also includes a section on the diagnostic imaging of vascular diseases of the liver and their treatment from a haematological standpoint (study of thrombotic diathesis and anticoagulant therapy). All recommendations are based on published studies extracted from “PubMed”. The quality of evidence and the strength of the recommendations were graded in accordance with the GRADE system (Grading of Recommendations, Assessment, Development and Evaluation). The evidence grade was classified into three quality levels: high (A), moderate (B) or low (C). The intensity of the recommendation was classified into two levels: strong (1) or weak (2) (Table 1). In the absence of sufficient evidence, recommendations were based on the opinion of the committee that wrote the guide.
Grades of evidence and recommendation (adapted from the GRADE system).
| Description | Symbol | |
|---|---|---|
| Grades of evidence | ||
| High | Further research is very unlikely to change our confidence in the estimate of effect | A |
| Moderate | Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate | B |
| Low or very low | Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Any estimate of effect is uncertain | C |
| Grade of recommendation | ||
| Strong | The existing evidence indicates that the benefits outweigh the risks and costs or vice versa. Therefore, the recommendation applies to the majority of patients. | 1 |
| Weak | The recommendation is made with less certainty: there is a greater cost or consumption of resources and the balance between these and the benefits is more fragile or uncertain. Any alternative could be equally reasonable | 2 |
Budd–Chiari syndrome (BCS) is defined as obstruction of hepatic venous outflow. This obstruction can be located anywhere from the small liver venules to the entrance of the inferior vena cava to the right atrium. This definition does not include obstruction caused by heart disease, pericardial disease or sinusoidal obstruction syndrome (previously called veno-occlusive disease), since these conditions have specific behaviours and prognosis.1 BCS is classified as primary when the obstruction is due to a primary process (vein thrombosis) and as secondary when there is compression or invasion of suprahepatic veins and/or of the inferior vena cava due to a lesion originating outside of the veins (e.g., a neoplasm). The object of these recommendations is primary BCS.
EpidemiologyThere is wide geographic variability in incidence of BCS. An annual incidence of 0.36 cases per million inhabitants has been estimated in France, 0.8 in Sweden and 0.13 in Japan.2–4 Studies suggest that in Western countries BCS is more common in women between 20 and 40 years old.5,6 However, in Asia there is a discrete predominance in men, with an average age of 45 at the time of diagnosis. In the West, isolated obstruction of suprahepatic veins is more common, while in Asia, combined obstruction of the inferior vena cava and the suprahepatic veins is more prevalent.6,7
One of the most extensive series published in the West included 237 patients with BCS diagnosed between 1984 and 2001.7 Two-thirds of patients were women and the mean age was 35 years (ranging from 13 to 76 years old). The most common location of the obstruction was in the suprahepatic veins (62%), followed by the inferior vena cava (7%) or both (31%); 34 patients (14%) also had portal vein thrombosis.
SymptomsThe clinical presentation of BCS is diverse and can vary from symptoms of acute liver failure with jaundice and hepatic encephalopathy to asymptomatic.6 The symptoms depend on the extent and speed of the onset of the thrombosis. The intensity and pattern of changes in transaminases and other liver function parameters also vary. In an extensive, prospective multicentre study, at the time of diagnosis, 83% of patients had ascites, 67% had hepatomegaly, 61% had abdominal pain, 58% had oesophagastric varices and 5% had gastrointestinal bleeding.8
In approximately 5% of patients, BCS can present as fulminant.1,8–11 In acute non-fulminant forms, BCS usually manifests with abdominal pain and severe ascites, aminotransferases can vary from 100–200IU/ml to more than 600IU/ml and alkaline phosphatase varies between 2 and 3 times normal values. Bilirubin does not tend to increase to levels that cause jaundice. In subacute forms, the start is subtle and manifests with ascites and minor hepatic necrosis. In chronic BCS patients are diagnosed due to the presence of clinical signs of chronic liver disease and complications of PHT. These patients may have normal or slightly elevated levels of serum aminotransferases, alkaline phosphatase and bilirubin.12 They may also present hypoalbuminaemia. Hepatopulmonary syndrome has been reported in more than 28% of these patients, if looked for intentionally.13
The concentration of proteins in ascitic fluid is variable, and although it is usually higher than in cirrhosis (between 1.5 and 4.9g/dl),13 the serum-ascites albumin gradient is elevated (>1.1).
During evolution, patients with chronic BCS may develop hepatocellular carcinoma at an accumulated incidence of 4% after a mean follow-up of 5 years.14
DiagnosisGiven the great heterogeneity in presentation, a diagnosis of BCS should be considered in any patient with acute liver failure, acute hepatitis or chronic liver disease.
Usually the diagnosis of BCS can be established noninvasively via Doppler ultrasound, which has a diagnostic sensitivity greater than 75%.15 The permeability of the splenoportal axis should also be assessed, since portal or splenic vein thrombosis may coexist in more than 10% of cases. If an experienced sonographer is not available, CT or magnetic resonance may be used to confirm the diagnosis and plan treatment. These two examinations can also be done when the ultrasound is apparently normal but BCS is highly suspected.15 The possible findings in these different techniques are described in the corresponding chapter.
If the non-invasive examinations are negative or not diagnostic, but there is a strong clinical suspicion of BCS, hepatic venography can help in the diagnosis by confirming the image of venous obstruction with or without the typical image of a “spider web” pattern of intrahepatic collateral veins.
A liver biopsy is not necessary to diagnose BCS. Nevertheless, it may be the only method to establish a diagnosis in cases in which only the small liver veins are thrombosed. A liver biopsy can also help determine the most appropriate treatment when liver failure is present, since if it is due to liver congestion, the patient may benefit from a shunt technique, while if the liver failure is due to cirrhosis, the patient will benefit from a liver transplant.16,17 The risk of bleeding should be taken into account in these patients, particularly if they are on anticoagulant therapy. This risk is lower if a transjugular liver biopsy is performed.
1.1.1Aetiological study in Budd–Chiari syndromeIn more than 80% of patients with BCS, an underlying disease is identified,8,13,18–23 and one quarter of these patients have more than one thrombotic risk factor.
Up to 50% of patients are diagnosed with a myeloproliferative neoplasm (polycythaemia vera, essential thrombocythaemia or primary myelofibrosis).7 The diagnosis of these conditions, in many cases, is not evident from peripheral blood smears due to the presence of PHT, splenomegaly and hypersplenism that mask the expected increases in complete blood count parameters. However, the possibility of determining the presence of the JAK2V617F mutation (present in 95% of patients with polycythaemia vera and in approximately 50% of patients with essential thrombocythaemia or primary myelofibrosis) has facilitated its diagnosis.24,25 Recently the presence of calreticulin (CALR) mutations in patients with splanchnic vein thrombosis, positive in 0.7–1.9% of patients, has been published.26–29 The JAK2 and CALR mutations are mutually exclusive.
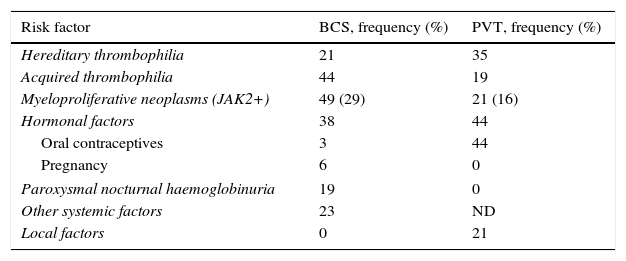
Approximately 20% of cases of BCS occur in women who are taking oral contraceptives, are pregnant, or have given birth within the last 2 months.30–34 In many of these patients, an exhaustive study of a potential thrombophilic cause highlights a specific state of hypercoagulability such that pregnancy or the use of contraceptives would act as a trigger. Table 2 summarises the prothrombotic conditions that have been described as causes of BCS and its prevalence.
Aetiological factors in Budd–Chiari syndrome and in portal vein thrombosis.
| Risk factor | BCS, frequency (%) | PVT, frequency (%) |
|---|---|---|
| Hereditary thrombophilia | 21 | 35 |
| Acquired thrombophilia | 44 | 19 |
| Myeloproliferative neoplasms (JAK2+) | 49 (29) | 21 (16) |
| Hormonal factors | 38 | 44 |
| Oral contraceptives | 3 | 44 |
| Pregnancy | 6 | 0 |
| Paroxysmal nocturnal haemoglobinuria | 19 | 0 |
| Other systemic factors | 23 | ND |
| Local factors | 0 | 21 |
BCS: Budd–Chiari syndrome; PVT: portal vein thrombosis; ND: no data.
Untreated symptomatic BCS has a mortality rate around 90% after 3 years of follow-up.35 The most common cause of death is refractory ascites with malnutrition, gastrointestinal bleeding and liver failure. Today, with the introduction of new treatment strategies, the prognosis has improved significantly. In a recent series that included 163 patients with BCS treated using a stepped-care therapy strategy from least to most aggressive, survival at 1, 2 and 5 years was 87%, 82% and 74%, respectively.8
Different parameters or combinations of parameters have been described to predict the prognosis of patients with BCS.7,35–37 Despite the fact that all these indices are valid for the evaluation of transplant-free survival and invasive therapy-free survival, their predictive precision for a specific patient is not sufficiently high to be useful in personalising treatment.38 Furthermore, the development of hepatocellular carcinoma or haematological disease progression can modify the prognosis of BCS.
TreatmentThere are no randomised studies that compare different therapeutic strategies in patients with BCS. Therefore, the recommendations are based on retrospective studies and clinical experience. The therapeutic strategy has changed over the years. Prior to 1990, the treatment was either medical or surgical; today, treatments with interventional radiology techniques have very high efficacy and are therefore the most commonly used. Currently stepped-care therapy is recommended, starting with medical treatment and progressing to more invasive strategies in the case of lack of response to the previous treatment.39–41
It is important to note that there are several objectives of BCS treatment: (1) to recognise and treat the underlying prothrombotic state; (2) to alleviate liver congestion to minimise impact on liver function and the onset of PHT, and (3) to treat the complications derived from developing PHT.
1.1.2AnticoagulationAnticoagulant therapy must be started as soon as possible and continue for an indefinite period to prevent the appearance of new re-thrombosis phenomena. Usually acenocoumarol or warfarin is used due to the ease of oral administration, and reaching a sustained international normalised ratio (INR) of 2–3 is recommended. Anticoagulation should be carried out even in asymptomatic patients and independent of other specific treatments for the underlying prothrombotic disease.41
1.1.3Correction of the flow obstructionChemical thrombolysisAttempts to recanalise the thrombosed vein. Experience with this technique is very limited and has only been proposed for patients diagnosed within the first 72h of evolution. Tissue plasminogen activator and streptokinase are the most widely used agents. It can be done locally or systemically. The local method allows for greater concentration of the thrombolytic substance in the thrombus area, but the side effects are similar to when the systemic method is used. This technique should be avoided if any invasive procedure has been performed, including paracentesis, in the previous 24h.42,43
Angioplasty with or without placement of a prosthesisIn patients with segmental, partial and short stenosis of the suprahepatic veins, transvenous or percutaneous angioplasty is indicated to attempt to re-establish physiological drainage of the veins.44,45 The main disadvantage is that restenosis frequently occurs, requiring new dilations, which may be avoided by placing expandable prosthesis.
Shunt treatment- (a)
Surgery. The intention of surgery is to convert the portal vein into a conduit for draining the congested liver. The two possible techniques are latero-lateral portacaval anastomosis and mesocaval anastomosis (not termino-lateral portacaval anastomosis, since this does not drain the liver). Mesocaval anastomosis is usually preferred since it is the simplest technique in the event of hypertrophy of the caudate lobe. When this compression is relevant (it is suggested when it creates a pressure gradient higher than 20mmHg), anastomosis is not effective. In these cases, in the era prior to implantation of a transjugular intrahepatic portosystemic shunt (TIPS), the placement of an expandable prosthesis had been proposed in the vena cava at the level of the compression and/or creation of a direct anastomosis between the mesenteric vein and the atrium (mesoatrial) There are little data about the efficacy of surgery in BCS, but it seems that it does not increase survival, due to high perioperative mortality (around 25%) and the high risk of shunt thrombosis. However, in patients who surpass the acute phase, the prognosis with surgery is usually excellent.46–48 Today, surgery has been relegated to those rare cases in which endovascular treatments are not possible.
- (b)
Transjugular intrahepatic portosystemic shunt (TIPS). TIPS is currently the shunt treatment of choice in BCS.36,49,50 Up to one-third of patients with BCS end up needing a TIPS.8 It offers clear advantages over surgery for its lower morbidity and mortality and due to the lesser technical complexity since, in expert hands, it can be performed in up to 90% of cases. In 45% of cases, no suprahepatic vein could be cannulated and a portal branch must be directly accessed from the intrahepatic vena cava. TIPS also allows for one of the obstacles to surgery, hypertrophy of the caudate lobe, to be overcome. Coated prosthesis should be used, since they decrease the risk of re-thrombosis and dysfunction.
In patients where shunting is not possible, is contraindicated, or cirrhosis is present with or without severe liver failure, the possibility of a liver transplant should be evaluated.51 Although a recurrence of the illness can occur in up to 11%, this is exceptional if anticoagulation is restarted early after the transplant. Also, there are prothrombotic haemostasis disorders that, because they are liver synthesis factors, disappear after transplant.
Despite the fact that in various studies the validity of a stepped-care treatment for BCS (Fig. 1) has been demonstrated,38,51 it has been suggested that a more aggressive treatment can be administered in symptomatic patients from the beginning, in order to avoid liver disease progression due to venous congestion. Nevertheless, currently there are no solid data that support said suggestion.
Monitoring
Patients with BCS must be monitored to rule out the onset of complications derived from disease progression, including oesophagastric varices or hepatocellular carcinoma, with a periodicity equal to that in patients with cirrhosis.12 Also, the possibility of transformation of the underlying myeloproliferative neoplasm must be monitored.
Recommendations- •
A diagnosis of BCS should be considered in the case of any patient with acute or chronic liver disease, whether it is symptomatic or asymptomatic (A1).
- •
Doppler ultrasound is the diagnostic method of choice. MRA and CTA can be used to confirm the diagnosis, especially if the ultrasound is not diagnostic and the degree of suspicion of disease is high (A1).
- •
We recommend referring patients with BCS to reference centres for diagnosis and treatment (A1).
- •
The presence of prothrombotic factors should be investigated in all patients with BCS. Identification of an isolated factor does not rule out the possibility of the existence of others (A1). To rule out the existence of a chronic myeloproliferative neoplasm (CMN), determining the JAK2V617F mutation is recommended (A1). If this is negative, it is recommended to determine the calreticulin gene. If the patient has a CMN or one is suspected, the patient should be referred to a haematologist (B2).
- •
The specific predisposing prothrombotic factor should be treated (B1).
- •
In patients with BCS in the cirrhotic phase, complications of PHT should be treated (C2).
- •
Anticoagulant treatment is recommended in all patients with BCS (A1). Complications deriving from PHT, if correctly treated, do not contraindicate anticoagulant therapy (B1).
- •
Temporary suspension of anticoagulants should be considered prior to any invasive procedure, including paracentesis (B1).
- •
Considering angioplasty with or without placement of a prosthesis is recommended as a first-line decompression procedure in patients with short stenosis of the suprahepatic veins or the inferior vena cava (A1).
- •
Patients who do not respond to initial medical treatment or to angioplasty with or without prosthesis should be treated with shunt techniques (A1). Close monitoring should be carried out for early detection of the failure of these treatments. The shunt treatment of choice is TIPS with a coated prosthesis (A1). If the TIPS fails or cannot be performed, considering surgical shunting is suggested (B1).
- •
Liver transplant is a rescue treatment in patients for whom shunt treatments have failed (A1), both in situations of acute and chronic liver failure. Many patients should continue with anticoagulant therapy after a liver transplant.
- •
Patients with BCS should be monitored to rule out the appearance of hepatocellular carcinoma. Given that differentiating between benign and malignant nodes does not tend to be easy and may require biopsy, it is recommended to do so at reference centres (A1).
The diagnosis of idiopathic portal hypertension (IPHT) must be considered when PHT exists in the absence of cirrhosis and other causes of non-cirrhotic PHT have been ruled out.52
The nomenclature of this condition has been very ambiguous and has hindered the advancement of knowledge of its aetiopathogenesis. For years it has received different names, such as hepatoportal sclerosis, non-cirrhotic portal fibrosis, IPHT, incomplete septal cirrhosis and nodular regenerative hyperplasia. It has been suggested that the symptoms of IPHT would be the symptomatic tip of the iceberg in which a significantly higher number of asymptomatic patients would have histological lesions consistent with hepatoportal sclerosis in which a significant number of these patients would never develop the clinical symptoms.
The epidemiology of this condition is unknown; however, it predominates in males, with an average age of onset of 40 years, although it has also been exceptionally described in children.53–55
The main aetiological factors identified associated with the development of IPHT are immune system disorders, infections, HIV and/or its treatments, drugs (azathioprine) and toxins, genetic predisposition and thrombophilia. In Western countries, a prevalence of underlying thrombophilic disorder has been described in up to 40% of patients with IPHT.52,53 A genetic basis for the disease has been postulated. The presence of mutations in the DGUOK56 and KCNN357 genes has been reported recently in patients with IPHT; however, more information is needed to establish a causal association.
SymptomsAt the time of diagnosis of IPHT, most patients have signs or complications of PHT.52 Haemorrhage secondary to PHT, mainly due to oesophageal varices, is the most common initial manifestation.58 Splenomegaly is a commonly-observed symptom in patients with IPHT, more common than in other causes of PHT, such as cirrhosis and portal vein thrombosis.53 Usually patients have conserved liver function at the time of diagnosis; only a minority have deterioration of liver function and this usually occurs in the context of intercurrent processes. The onset of ascites, although rare, is a factor of poor prognosis.52 The onset of hepatic encephalopathy is very uncommon, and usually is due to the presence of large portosystemic collaterals.59 A recent Spanish study that evaluated 69 patients with IPHT found that 58% were asymptomatic at the time of diagnosis, with the detection of thrombocytopenia and splenomegaly being the main motives for the diagnosis of IPHT.60
DiagnosisThere is not currently a diagnostic test that can be considered the gold standard for diagnosing IPHT, which rules out other causes of PHT (Table 3). Frequently, patients with IPHT are erroneously deemed to have cirrhosis, due to the presence of a nodular liver in imaging tests along with signs of PHT.53,61 Conducting a liver elastography can be useful for a suspected diagnosis of IPHT. A low liver elastography value (<12kPa) in a patient with clear signs of PHT should make us suspect a diagnosis of IPHT.52,62 A recent Spanish study has identified metabolomic analysis of serum samples as a potential tool to help diagnose IPHT.63
Diagnostic criteria for idiopathic portal vein hypertension.
| 1. Presence of clinical symptoms of portal vein hypertension |
| Splenomegaly/hypersplenisma |
| Oesophageal varices |
| Ascites |
| Slightly elevated venous pressure gradient |
| Presence of portal collaterals |
| 2. Rule out cirrhosis via liver biopsyb |
| 3. Rule out chronic liver diseases that cause cirrhosis or non-cirrhotic portal hypertension |
| Chronic hepatitis due to hepatitis B or C |
| Nonalcoholic steatohepatitis/alcoholic steatohepatitis |
| Autoimmune hepatitis |
| Hereditary haemochromatosis |
| Wilson disease |
| Primary biliary cholangitis/primary sclerosing cholangitis |
| 4. Rule out diseases that cause non-cirrhotic portal hypertension |
| Congenital hepatic fibrosis |
| Sarcoidosis |
| Schistosomiasis |
| 5. Permeable portal and hepatic veins (via Doppler ultrasound or abdominal CT) |
To diagnose IPHT, all criteria must be met. When all criteria except number 5 are met, but there are data in the biopsy characteristically associated with IPHT, it is considered probable IPHT.
Today, liver biopsy continues to be essential to establish a diagnosis of IPHT. The crucial factor is that it rules out the presence of cirrhosis. Also, reviewed by expert pathologists, there are some abnormalities that, although not pathognomonic, support the diagnosis. These include phlebosclerosis, nodular regeneration, sinusoidal dilatation, paraportal venous anastomosis and perisinusoidal fibrosis.53,61 It has been suggested that phlebosclerosis is the initial lesion that causes intrahepatic haemodynamic changes. Thus, the possible obliteration of portal venules would condition a change in intrahepatic circulation and, as a result, a remodelling of the parenchyma (nodular regeneration) with hepatocyte atrophy of the areas with decreased venous flow and compensatory hyperplasia in the better perfused areas.
To reach the correct diagnosis, it is important to obtain a high-quality, large liver biopsy to be able to detect these changes, which are sometimes minimal.
Natural historyMortality due to PHT complications, mainly variceal haemorrhage, in patients with IPHT is significantly less than what is reported in cirrhotic patients,52,53 probably due to the fact that the majority of patients with IPHT conserve their liver function. Nevertheless, in cases in which progressive deterioration of liver function is shown, liver transplant may become necessary.59,61 It is postulated that unfavourable disease evolution can involve a precipitating factor or an additional cause of liver damage.53 Deterioration of liver function and the appearance of ascites can be explained by a reduction in portal flow and, consequently, atrophy of the peripheral liver parenchyma.52
A high incidence of portal vein thrombosis has been reported in patients with IPHT.53 In these cases, starting anticoagulant therapy early is capable of achieving recanalisation in up to 54% of patients.60
Patients with IPHT frequently develop liver nodules, mostly benign; however, it must be kept in mind that the appearance of hepatocellular carcinoma has also been anecdotally reported. Currently, and according to available data, the screening for hepatocellular carcinoma cannot be recommended routinely in patients with IPHT.52 Although a nodule in these patients can present the radiological characteristics of hepatocellular carcinoma, this diagnosis cannot be established, since these criteria are only validated in cirrhosis of the liver.
In general, mortality due to liver causes is low, and the impact on the reduction of survival in patients with IPHT is usually due to high mortality of the underlying associated disorders.61
Treatment1.1.4Prophylaxis for gastrointestinal haemorrhageThe current recommendation is to manage these patients following the same recommendations as for patients with cirrhosis, although there are little specific data on patients with IPHT.
Endoscopic treatmentEndoscopic treatment is effective in controlling acute haemorrhage in 95% of cases64 and as secondary prophylaxis in patients with IPHT.65 There are no data on the efficacy of variceal ligation in these patients. However, the superiority of this treatment compared to variceal sclerosis in cirrhotic patients supports the use of ligation in patients with IPHT and varices.
Non-selective beta blockersDespite the absence of specific data about the efficacy of non-selective beta blockers in patients with IPHT, the great evidence of its benefit in patients with cirrhosis justifies its use in IPHT.
Portosystemic shuntTIPS has recently been shown to be an excellent option for the treatment of refractory haemorrhage in patients with IPHT.66 Although uncommon due to the conserved liver function in the majority of cases, shunt treatment increases the risk of onset of hepatic encephalopathy, above all in the subgroup of patients with portal vein thrombosis.66 Surgical portosystemic anastomosis is a safe strategy in patients with IPHT,67 but TIPS, due to its lesser invasiveness, should be considered as the first-choice option.
1.1.5AnticoagulationThere are insufficient data that support the use of anticoagulant therapy to prevent thrombosis in patients with IPHT. The current recommendation is relegated to patients who develop portal vein thrombosis and/or have a clear underlying prothrombotic abnormality.
1.1.6Liver transplantIn patients with progressive deterioration of liver function and untreatable complications of PHT, liver transplant should be considered.53,59,61
Recommendations- •
A diagnosis of IPHT should be considered in patients with signs of PHT once cirrhosis and other specific causes of PHT have been ruled out (B1).
- •
A liver biopsy is necessary to diagnose IPHT (A1).
- •
Management of PHT complications should follow the recommendations established for cirrhosis (B1).
- •
Evaluating the possible development of portal vein thrombosis every 6 months is recommended (B1).
- •
Patients with IPHT can develop liver nodules that mainly, but not always, are benign (C).
- •
Liver transplant can be considered in patients with IPHT who develop liver failure or untreatable complications of PHT (B1).
Portal vein thrombosis (PVT) is defined as the presence of thrombosis in the portal vein trunk or in the intrahepatic branches that can extend to the splenic vein, the mesenteric vein or both. When a cavernoma has already formed, it is called chronic PVT, being an evolutionary phase of the same condition. The incidence of PVT of any aetiology is 0.7 in 100,000 inhabitants/year, and the prevalence is 3.5 per 100,000 inhabitants,68 constituting the primary cause of pre-hepatic PHT.69
Symptoms and natural historyThe symptoms of PVT vary whether it is diagnosed in the acute or chronic phase and depend on the velocity of formation of the thrombosis, the spreading thereof and the development of secondary complications.
1.1.7Acute portal thrombosisThe principal symptom is abdominal pain (90–95%), with a very variable presentation, encompassing from nonspecific symptoms of dyspepsia to the onset of acute abdomen, including with intestinal ischaemia, when the mesenteric veins are affected. This is the primary cause of death. Therefore, in the case of symptoms of intense abdominal pain, in the absence of peritoneal irritation, occasionally with diarrhoea and rectorrhagia, with suggestive analytical data (metabolic acidosis, increase in LDH and/or lactate) and kidney or respiratory failure, the study should be completed with imaging techniques (CTA) and an early surgical assessment should be requested.70–73 Other possible symptoms are fever (approximately 50%), occasionally with systemic inflammatory response syndrome in the absence of sepsis, and ascites (35%), which are usually few, usually detected in imaging tests. Finally, there is a non-negligible proportion of patients in whom PVT develops asymptomatically or with nonspecific symptoms in which the acute episode goes unnoticed, and they are diagnosed in the portal cavernoma stage.1,74,75
Acute septic PVT (pylephlebitis) usually presents as a febrile syndrome, with discomfort, nausea, abdominal pain and occasionally shock with or without liver abscesses, that appear associated with the causal abdominal infection symptoms: appendicitis, diverticulitis, colitis, cholangitis, cholecystitis or cholecystopancreatitis. It is essential to obtain haemocultures, which are positive in 45–77%. The presence of portal gas is a specific, although uncommon, radiological finding.76–78
1.1.8Chronic portal thrombosisIn this stage the patient usually remains asymptomatic. When symptoms appear, they arise from the development of PHT or the existence of portal cholangiopathy. The symptom most frequently associated with PHT is haemorrhage due to oesophagastric varices, which is usually better tolerated than in other forms of PHT, probably due to the good liver function. The predictive factors for bleeding are the size of the varices and the prior history of bleeding.1,74 Iron deficiency anaemia or bleeding can appear due to gastropathy from PHT or ectopic varices (duodenal, anorectal, perivesicular, biliary tree) which are more common than in other causes of PHT, but in which bleeding is rare.74,79 Ascites, when it appears, is usually easy to treat75,78,80 and is usually associated with triggering factors such as upper gastrointestinal haemorrhage or infection. Hepatic encephalopathy has been reported very infrequently,74 with the possible presence of minimal hepatic encephalopathy.81
DiagnosisDoppler ultrasound is the first test to be carried out in case of suspected PVT. It should be performed by experts, since this technique depends on the examiner. Its sensitivity is 89–94% and its specificity is 92–96%.82–84 The findings of this exam are indicated in the radiology section. It is vital to perform a CTA and/or an MRA at the time of diagnosis to evaluate the extent of the thrombosis and study its possible aetiology. These techniques should also be used to reach the diagnosis of PVT in patients with high clinical suspicion and the absence of consistent ultrasound data.1,41,72,74,85–87 It should be kept in mind that acute re-thrombosis phenomena may occur in patients with known portal cavernomas, which are usually harder to diagnose.
Ascites may appear in the acute phase, although in this case it is part of the inflammatory process of acute PVT, and therefore tends to be little and transient.80 Splenomegaly may be secondary to established PHT or a chronic myeloproliferative neoplasm as causal prothrombotic pathology.73,75,78,80
Liver function tests are usually normal or slightly altered temporarily.1,74,79,88
1.1.9Aetiological diagnosis of portal vein thrombosisThe aetiological diagnosis of PVT is essential, since it can change the therapeutic approach. Causes include acquired or hereditary thrombophilic factors, local factors and other possible triggers that are less common in our environment.1,41,75,79,88–92 In 75–81% of cases, it is possible to find at least one prothrombotic factor, and in 18–36%, the presence of two factors has been reported; thus, only 18–25% of cases are considered idiopathic.75,91,92 Recently, central obesity has been included as a factor associated with idiopathic PVT.93
Therefore, it is necessary to conduct an exhaustive aetiological study with the collaboration of a haematology specialist, including the detailed evaluation of personal and family history of venous thromboembolism phenomena, reported in 14% and 24% of cases, respectively.75
Myeloproliferative disorders are the most commonly-described cause: the evaluation of peripheral blood can be conditioned by haemodilution and hypersplenism associated with the presence of PHT. Therefore, the JAK2V617F gene mutation must be systematically requested. If negative, study the somatic mutation of the calreticulin (CALR) gene and, if both are negative, the bone marrow scores should be assessed, in addition to considering repeating the test as the condition evolves (Table 2).1,41,75,79,88–93 In children, the congenital origin of portal vein cavernoma has been assessed as a possible aetiology.74
1.1.10Diagnosis of portal vein thrombosis complicationsOesophagastric varices and portal hypertension gastropathyA recently-published study evaluated the natural history of oesophagastric varices in patients with non-cirrhotic PVT. This study found that at the time of diagnosis of PVT, 66% of patients have oesophagastric varices (19% small oesophageal varices, 40% large oesophageal varices and 7% isolated gastric varices). In the 34% of patients without oesophageal varices, the probability of developing them is 2%, 22% and 22% at 1, 3 and 5 years, respectively. The presence of ascites and splenomegaly were identified as independent risk factors for developing varices. The probability of growth of small oesophageal varices at the time of diagnosis was 13%, 40% and 54% at 1, 3 and 5 years, while the probability of haemorrhage due to oesophagastric varices in patients on primary prophylaxis was 9%, 20% and 32% at 1, 3 and 5 years. Finally, mortality secondary to haemorrhage due to oesophagastric varices was 0.7%. The study concludes that the evolution of varices in non-cirrhotic PVT is similar to that in patients with cirrhosis, which supports conducting the same follow-up in both conditions.94 Oesophagastric varices can form starting from the first month after diagnosis of acute symptomatic PVT80; therefore, an upper digestive tract endoscopy should be performed early (2–3 months from the PVT episode), and should be repeated at 6–9 months if there are no varices or they are small, especially in the absence of recanalisation. Later, the schedule usually used for cirrhosis will be followed, with endoscopy every 2–3 years in patients without varices and every 1–2 years in patients with small varices.1,72,87,94
Portal cholangiopathyThe pathogenesis of portal cholangiopathy (PC) is not completely clear. Some authors indicate that chronic PVT and cavernous transformation cause ischaemia in the bile ducts, although the most accepted theory states that periportal collaterals that make up the cavernoma can exert sufficient pressure on the bile duct to cause symptoms of cholestasis and, on occasion, true stenosis of the bile duct. Most cases of PC are associated with chronic extrahepatic PVT (81–100%), although this condition can also appear in patients with marked PHT without thrombosis, whether of cirrhotic (0–33%) or idiopathic (9–40%) origin.95 Regarding the symptoms, most patients are asymptomatic. In the largest series of PC cases published to date, which included 67 patients, only 21% had symptoms, with the most common being abdominal pain, jaundice and acute cholangitis. The authors of this paper propose a classification of PC based on the radiological findings (degree I: bile duct angulation; degree II: stenosis without dilatation; degree III, stenosis with dilatation) and they observe that only cases with stenosis and bile duct dilatation (degree III) have symptoms. The mean time elapsed between the PVT episode and onset of symptoms related to PC was 42 and 118 months in cases of acute and chronic PVT, respectively. There are cases described in the medical literature with latency periods exceeding 10 years.96
MR cholangiography is the technique of choice for diagnosis and classification of this condition.87,96,97 There are two classifications of PC: one anatomical, focused on the type of bile duct lesion,98 and another that shows a clinical–radiological correlation which allows morphological characteristics to be combined with symptoms, since only patients with degree III lesions have bile duct complications.96 In our environment, given the results of this Spanish series, it is recommended that if there is no degree III PC present on the initial MR cholangiogram, portal repermeation does not occur and no re-thrombosis phenomena are present, the examination should be repeated at 12 months from the initial test, without the need for additional examinations due to the precocity of this condition after the formation of PVT and the low probability of progression after this period.87,89,96
TreatmentThe objective of treatment for PVT is to revert or prevent progression of thrombosis in the portal venous system and to treat complications. It is also essential to treat the underlying associated disease, if any. Most decisions should be individualised, depending on the local experience, given that there are no controlled clinical trials.
1.1.11Acute portal thrombosisAnticoagulationStudies that have evaluated the efficacy of anticoagulant therapy in acute PVT indicate that complete and partial recanalisation is achieved in around 40% and 22%, respectively.75,80 The possibility of recanalisation rises to 69% if the anticoagulant therapy is started in the first week after the onset of symptoms, in comparison with 25% if started late,80 with spontaneous repermeation being uncommon.75,78,99 For this reason, anticoagulant therapy should be started early in all patients, except when contraindicated.75,78,80 It is important to note that recanalisation of the portal vein not only prevents the development of PHT,80,100 with the consequential impact on prognosis, but also reduces the risk of serious complications, such as peritonitis from intestinal necrosis due to ischaemia.80 The factors predictive of recanalisation failure despite anticoagulant therapy are the presence of ascites (including degree 1), the spread of thrombosis to the splenic vein, the presence of various prothrombotic factors and delayed start of anticoagulants.75,80 The duration of anticoagulant therapy is not well defined. However, in a recent multicentre study, recanalisation was observed in the portal vein at least up to 6 months after having started treatment.75 This study also observed that when the thrombosis affects the splenic vein and/or the superior mesenteric vein, recanalisation can be achieved up to 12 months after the start of anticoagulant therapy.75 For this reason, it is recommended to maintain anticoagulant therapy for at least 6 months when the portal vein trunk and/or its branches are affected, while in patients with mesenteric and splenic involvement, anticoagulants can be considered for up to 12 months. The incidence of haemorrhagic complications in anticoagulated patients is low and mortality due to anticoagulation is practically zero.75,78
Early start of low-molecular-weight heparin (LMWH) as an anticoagulant drug is recommended. For permanent anticoagulation, use of oral anticoagulants with vitamin K antagonists is recommended, with a therapeutic range defined by an INR value between 2 and 3.72
ThrombolysisThrombolytic treatment via a catheter inserted into the portal vein transhepatically has been evaluated in no more than 100 patients, many of them published only as case reports. The probability of recanalisation is similar to that achieved with anticoagulation; however, the morbidity and mortality reported is high and haemorrhage at the puncture site is 60%.101–103 Transjugular access seems to have fewer complications, although the data are limited to fewer than 30 patients.104,105 The experience with surgical or mechanical thrombectomy and transjugular intrahepatic portosystemic shunting (TIPS) is very limited. Surgical thrombectomy is not recommended given the frequent recurrence of thrombosis and high morbidity and mortality.106 Experience with percutaneous mechanical thrombectomy or associated with TIPS is also very limited; furthermore, these techniques can cause vascular trauma or trauma to the inner layer of the portal vein and, therefore, be associated with recurrent thrombosis.107 Finally, there are no studies that have evaluated the risk-benefit ratio of these treatments compared to early anticoagulation. Keeping in mind as well that the long-term prognosis in patients with chronic PVT is generally good and that these techniques have high morbidity and mortality, their indication is very doubtful.
1.1.12Chronic portal thrombosisAnticoagulationAnticoagulation has a controversial role in chronic PVT. Its theoretical objectives are to avoid rethrombosis of the splenoportal axis, which could aggravate the PHT and to avoid thrombotic phenomena in other vascular areas (both arterial and venous).108,109
There are no controlled prospective studies that evaluate the risk-benefit ratio of anticoagulant therapy in the prevention of rethrombosis in patients with portal vein cavernomas, and all the information is based on retrospective cohorts in which no distinction was made between patients with and without prothrombotic risk factors. In one of these studies, anticoagulation significantly reduces the risk of new thrombotic episodes,109 while another only shows a tendency to reduce this risk, but not significantly.108 Regarding the risk of haemorrhage, there are conflicting data: in the first of the studies published, anticoagulant therapy did not increase the risk of haemorrhage or an increase in the severity thereof109; however, in the second study, anticoagulation was related, in a statistically significant manner, to the onset of haemorrhagic phenomena that, nevertheless, did not influence survival. Notably, survival was influenced by the appearance of new thrombotic episodes. Thus, this study suggests a possible beneficial effect of anticoagulation on survival, despite the observed increase in haemorrhagic risk.108
Despite the relative lack of data currently available, permanent anticoagulant therapy is considered indicated in patients with an underlying prothrombotic disorder that so requires it, and should be considered in patients with recurrent thrombotic episodes, family history of deep vein thrombosis and history of intestinal ischaemia at the time of the acute episode.41,72,74,108,109 It is important to emphasise that the start of anticoagulant therapy should be preceded by adequate primary or secondary prophylaxis for upper gastrointestinal haemorrhage due to oesophagastric varices.41,72,74,109
Treatment of portal vein hypertensionIn patients with non-cirrhotic chronic PVT, the recommendations for prophylaxis and treatment of haemorrhage due to oesophagastric varices are based on the studies conducted in patients with cirrhosis and PHT.72 This is due to the fact that there are no controlled studies that have evaluated treatment of variceal haemorrhage in this context. Also, starting primary prophylaxis with non-selective beta-blockers or endoscopic treatment with ligation is recommended in patients with large oesophageal varices (>5mm).72
Regarding secondary prophylaxis, it has also been shown that endoscopic removal of oesophageal varices in patients with non-cirrhotic PVT significantly decreases the risk of rebleeding and the combined use of beta-blockers and endoscopic ligation treatment is accepted.72
There are also no studies evaluating which treatment is best to control an acute episode. The latest consensus from Baveno VI recommends using vasoconstrictors and/or endoscopic treatments.72 In the case of a failure of medical or endoscopic treatment, a TIPS could be considered. Placement success rates vary between 75% and 100%, according to one study, with a hepatic encephalopathy rate similar to that of patients with cirrhosis.110 However, a recent study shows a lower chance of success in placement (less than 35%), but with high efficacy in controlling haemorrhage in the group that achieved a functioning TIPS (69% vs 14%; p=0.057).111 Therefore, more studies, with a wide series of patients, are needed to evaluate the real role of TIPS in this indication. Also, there is a possibility of attempting portal repermeation using interventional angiography-radiology techniques with the possible placement of prosthesis.112 In the case of being unable to perform a TIPS, and if a surgical rescue shunt anastomosis is considered, it should be carefully analysed whether there are suitable permeable vessels for a possible shunt. In fact, in up to 50% of patients with PVT, splenic and upper mesenteric vein thrombosis are also observed,75,90 facts that preclude shunt surgery. In addition, if there is any suitable vessel, each patient should be individually evaluated to determine whether the planned shunt is able to resolve the PHT symptoms a priori.
In the event of persistent or uncontrollable haemorrhage and the inability to perform one of the previously-mentioned techniques, other measures may be attempted such as selective surgical devascularisation (oesophageal transection),113 splenectomy or surgical ligation of the varices. In children with a permeable upper mesenteric vein and a permeable left portal branch, a shunt between these two veins may be inserted, which is known as a mesenteric Rex shunt.114,115 This technique will allow the portal vein system to decompress and also revascularise the liver with portal blood. There is no experience with this type of shunt in adults.
Treatment of portal cholangiopathyTreatment of portal cholangiopathy (PC) has not been evaluated in prospective, controlled studies. The treatment recommendations are therefore based on expert consensus. In asymptomatic patients with radiological PC, or merely with discrete increase of cholestatic enzymes, no type of treatment is recommended.41 Enzymatic disturbances usually resolve with the use of ursodeoxycholic acid, although prospective controlled studies that evaluate this alternative would be needed. A recently-published expert consensus recommends endoscopic treatment with sphincterotomy and bile duct drainage in symptomatic patients with bile duct obstruction with or without calculi.95 Bile duct drainage should be performed by placing a prosthesis in the common bile duct, which requires periodic replacement due to its frequent obstruction. Ursodeoxycholic acid may be beneficial in this context, but again, this strategy should be evaluated. Portal decompression via shunt surgery in the splenoportal axis or TIPS would be reserved for cases of recurrent symptoms despite endoscopic treatment.95,116 Bilioenteric shunts are associated with high morbidity and mortality and therefore are not recommended.95
PrognosisBetter knowledge of the disease and improvement in imaging techniques has allowed for early diagnosis of acute PVT, which allows for early starting of anticoagulation, with reported 5-year survival at 85%. Nevertheless, if there is an intestinal infarction and multiple organ failure, mortality can be 20–50%.79 In patients with chronic PVT, global mortality is less than 10%,117 although there is no specific comparison with the mortality observed in the general population. It is important to indicate that in more than 50% of cases, the cause of death is not directly related to the PVT. When this is responsible for the patient's death, in half of all cases it is due to gastrointestinal haemorrhage, with the rest of cases being associated with spread or recurrence of thrombosis.109 The factors predictive of survival have not been adequately studied, although the determining principles seem to be advanced age and the presence of mesenteric thrombosis.79
Recommendations- •
It is recommended to rule out acute PVT in any patient with acute persistent abdominal pain.
- •
It is recommended to rule out chronic PVT in patients with PHT without known liver disease.
- •
Doppler ultrasound is the first technique for diagnosing PVT. CTA and/or MRA are necessary to evaluate the extent of PVT.
- •
In light of a finding of PVT, the presence of underlying chronic liver disease should be ruled out.
- •
In acute PVT, an upper gastrointestinal endoscopy should be performed 2–3 months after diagnosis, which should be repeated 6–9 months if there are no varices or they are small. In chronic PVT, endoscopy should be performed at the time of diagnosis. Later, the recommendations for cirrhosis will be followed.
- •
In chronic PVT, conducting an MR cholangiogram during diagnosis to evaluate the presence of PC is suggested.
- •
In acute PVT, starting anticoagulant therapy early, and maintaining it for a minimum of 6–12 months is recommended. There is little experience to recommend other treatments such as thrombolysis, surgical thrombectomy or placement of a TIPS.
- •
If there is any prothrombotic risk factor, intestinal ischaemia during the episode, or family or personal history of venous thromboembolism, it is recommended to maintain anticoagulant therapy indefinitely to prevent rethrombosis.
- •
Prophylaxis and treatment of PHT complications should follow the recommendations established for cirrhosis.
The consideration of cirrhosis as a disease with a prohaemorrhagic phenotype has clearly changed in recent years. In addition, it has been shown that in patients with cirrhosis there is a prothrombotic tendency mainly due to a change in the complex physiological equilibrium between pro- and anticoagulant factors.118 This imbalance is clinically associated with an increase in the risk of thrombosis in different areas, including the lower limbs and pulmonary thromboembolism, and, most notably, splenoportal axis thrombosis (PVT).119,120
Furthermore, this physiopathological and clinical reality suggests the possibility of including anticoagulant therapy (heparin or vitamin K antagonists) among the possible therapeutic options in vein thrombosis in patients with cirrhosis, which has classically been considered a contraindication.
Incidence and prevalencePVT is the most frequently observed type of thrombosis in patients with cirrhosis, and its prevalence depends to a great extent on the population studied and the diagnostic procedure. Thus, prevalences that range from 2.1% to 23.3% have been reported in the different series collected in patients that are candidates for transplant without hepatocellular carcinoma.121 Remarkably, around half of the patients in which PVT has been detected at the time of transplant did not have a diagnosis prior to the surgery, which probably indicates an insufficient capacity to detect the problem. Moreover, various cohort studies have reported an annual estimated incidence of 4.6–17%.122–125 Finally, the presence of cirrhosis is associated with a 7.3% increase in relative risk for development of PVT compared to the general population.126
In short, all these data highlight the presence of a quantitatively relevant problem with potential clinical repercussions.
SymptomsIn the majority of cases, PVT in cirrhotic patients is asymptomatic and is detected casually in biannual monitoring ultrasounds, although it can occasionally be diagnosed along with liver decompensation. However, it has been suggested that the presence of PVT can have clinically-relevant consequences given that it has been associated, independently, with a greater risk of variceal haemorrhage, with the failure of endoscopic treatment in the control of acute haemorrhage, with the risk of relapse, and with an increase in mortality at 6 weeks (36% in patients with PVT vs 16% in patients without PVT).127–129 In addition, the risk of intestinal infarction, as well as the associated mortality, are greater in patients with spreading of the thrombus to the superior mesenteric vein.130 It has been suggested that the impact of PVT could be greater in the context of a liver transplant.131 Also, the presence of PVT has been associated with a significant increase in mortality at 30 days and at 1 year after transplant compared to patients without PVT.121 However, it is important to note that this excess mortality risk has only been reported in the presence of complete PVT.131
Risk factorsThe development of PVT is determined by a change in the physiological equilibrium that regulates haemostasis, such as one of the components of Virchow's triad. The aetiopathogenesis of PVT in cirrhosis is probably multifactorial. Thus, a reduced velocity in portal flow is associated with an increased risk of developing PVT.123 Furthermore, it is difficult to study the presence of thrombophilia in cirrhosis due to the nonspecific decrease in anticoagulant factors.132 Nevertheless, various cohorts have investigated the possible role of genetic mutations associated with thrombophilia; the mutation of the prothrombin gene G20210A is the abnormality most commonly associated with PVT.130,133,134 Moreover, the most advanced liver disease (Child–Pugh C), the presence of PHT complications, prior endoscopic treatment of oesophageal varices with sclerosis and age are other factors that have been suggested as risk factors for the development of PVT.121,124,125,135
DiagnosisDoppler ultrasound is the first-line method of choice for diagnosis, with an approximate sensitivity of 90% if the PVT is complete and 50% if it is partial.121 CT or MR have a greater capacity to evaluate the extent of the PVT to other splanchnic vessels. In cirrhotic patients, PVT can commonly be found associated with hepatocellular carcinoma portal invasion. In these cases, the elevation of serum alpha-fetoprotein levels, the increase in portal vein diameter, the enhancement of the thrombus in the arterial phase with the administration of contrast medium,136 arterialised flow detection by Doppler ultrasound137 or the conduct of a thrombus biopsy allow for correct diagnosis.
TreatmentThere is limited information available regarding the benefit of anticoagulant therapy in patients with cirrhosis and PVT. Spontaneous recanalisation has been reported in PVT mainly when it is partial in patients not on anticoagulant therapy124,138,139, but the probability of progression is higher (from 48% to 70% of patients) in a 2-year follow-up.138,140
A controlled and randomised study showed that the administration of enoxaparin 4000IU/day for one year prevents the development of PVT without increasing the risk of haemorrhagic complications.141 Five published cohorts122,140,142–144 evaluated the efficacy of anticoagulant therapy in a total of 163 anticoagulated patients, most with partial PVT and different anticoagulation regimens (low-molecular-weight heparins [LMWH] or vitamin K antagonists). The repermeation rate was 55–75% in an average time of 6 months. Starting anticoagulant therapy early seems to be the most important predictive factor in recanalisation,140,143,145 with the mean time between PVT diagnosis and the start of anticoagulation within 6 months being the best predictor of possible response.140 The presence of ascites has also been associated with a lesser response to anticoagulation.145 There are no specific recommendations regarding the duration of anticoagulant therapy, although a recurrence of PVT in up to 38% of patients has been reported upon discontinuing anticoagulants.143 Therefore, it is suggested that extending anticoagulant therapy after repermeation could prevent rethrombosis. Also, maintaining anticoagulant therapy indefinitely could be considered in patients with PVT progression, spreading of thrombosis to the superior mesenteric vein, history of intestinal ischaemia or the presence of any known procoagulant factor.41,72
A multicentre study showed a greater risk of haemorrhage in patients with a platelet count lower than 50×109/l.143 Another recent multicentre study evaluated the impact of anticoagulant therapy on patients with liver cirrhosis and upper gastrointestinal haemorrhage, with comorbidity and degree of multiple organ failure, not the anticoagulant therapy itself, being the factors affecting the morbidity and mortality of these patients.146
There are very preliminary data about the new direct anticoagulants that suggest that they could be an option in patients with cirrhosis, with an efficacy similar to traditional anticoagulants without increasing the risk of haemorrhage or hepatotoxicity.147–149 Nevertheless, there are insufficient data to recommend their use.
In any case, it is recommended to implement adequate prophylaxis for variceal haemorrhage, with beta blockers or endoscopic variceal ligation, before starting any type of anticoagulant therapy in these patients.1,41,72
Although the applicability and efficacy of TIPS to treat PVT are not well established, it is a possible option in patients with PVT, with it being technically possible even in some select cases with portal vein cavernomas.140,150–152 However, in most cases the indication for TIPS is not PVT itself but other complications of PHT. In the presence of intrahepatic portal branch thrombosis a percutaneous approach may be necessary, which is associated with an increase in the risk of complications during the procedure.153,154
Controlled and randomised studies are absolutely necessary in order to make recommendations about the best therapeutic strategy in this complex environment. While awaiting more robust data, every institution should set its own algorithm according to patient types and its own experience, taking into account important factors such as the possible indication of transplant, the degree and extent of the PVT, the existence of underlying prothrombotic factors, the history of prior thrombosis or the presence of PHT complications, among others.1,41,72
Recommendations- •
Evaluate the permeability of the portal vein in all patients with liver cirrhosis on the transplant waiting list or those who are potential candidates for it (B2).
- •
In light of an initial diagnosis of PVT, its extent should always be evaluated via CTA or MRA (A1).
- •
In patients with hepatocellular carcinoma and PVT, the possible tumoural nature should be ruled out via imaging tests with contrast or a thrombus biopsy (A1).
- •
In patients with cirrhosis and PVT, a study of prothrombotic factors can be considered (B2).
- •
In patients with PVT and oesophageal varices, adequate prophylaxis for haemorrhage due to PHT should be done prior to starting anticoagulation (A1).
- •
Starting anticoagulant therapy in patients with PVT on the liver transplant waiting list or those who are potential candidates for it, especially when there is spreading to the superior mesenteric vein or the thrombosis progresses during monitoring, is recommended.
- •
Anticoagulant therapy at therapeutic doses should be maintained for a minimum of 6 months (B1). In patients with a history of intestinal ischaemia, with known prothrombotic factors, thrombosis that progresses in monitoring or who are candidates for a liver transplant, maintaining anticoagulant therapy indefinitely or until the transplant can be considered (C2).
- •
In patients who are candidates for a liver transplant and who present PVT that progresses in monitoring without responding to anticoagulation, the placement of a TIPS can be considered (B2). In these cases it is important to adapt the length of the TIPS to avoid technical difficulties at the time of transplant.
Arterioportal fistula (APF) is an abnormal arteriovenous connection between any splanchnic artery and portal circulation. Normally, the most commonly-involved artery is the hepatic artery (65%), followed by the splenic (11%) and the superior mesenteric artery (10%). It is an uncommon cause of presinusoidal PHT. Although rare, its incidence is increasing due to the increase in invasive procedures at the hepatic level.155
APFs can be acquired or congenital. Acquired APFs (more common) are usually due to invasive procedures (percutaneous or transjugular liver biopsies, placements of TIPS, transparietohepatic biliary drainage, etc.), although they can also occur after trauma, due to the rupture of an aneurysmal artery in the portal system, related to cirrhosis, hepatocellular carcinoma or other benign tumours.156–158 According to their characteristics, the following classification has been proposed by Guzman et al.159
- •
Type 1. Small-size APFs, normally intrahepatic and peripheral, with hardly any functional repercussions. This tends to be the case in fistulas secondary to a liver biopsy. The patient normally appears asymptomatic and the fistula can resolve itself in a month due to a thrombosis mechanism.
- •
Type 2. Larger APFs, with sufficient flow to cause PHT, are usually extrahepatic or central intrahepatic and develop after a trauma or due to the rupture of an arterial aneurysm in the portal system.
- •
Type 3. Congenital APFs are less common (<10%). They tend to be intrahepatic, diffuse or multiple and are a cause of severe childhood PHT.160 The causes of congenital fistulas include Osler–Weber–Rendu syndrome, Ehlers–Danlos syndrome, arteriovenous malformations and aneurysms.161,162
The finding of an isolated fistula in adulthood is typically acquired.
PathophysiologyAPFs are a cause of presinusoidal PHT, mainly due to an increase in portal flow that increases at the expense of the arterial flow that feeds the fistula. In cases of long evolution, due to the arterialisation of the portal vein, intimal hyperplasia is also observed. The severity of the symptoms is related to the volume of blood that circulates through the fistula, while liver function stays relatively conserved, in the absence of prior liver disease.156
SymptomsMost cases are asymptomatic. Central and larger APFs, and APFs with higher flow, manifest as complications related to the PHT. The most common presentation is high or low gastrointestinal haemorrhage (33%), normally due to rupture of oesophageal varices, followed by ascites (26%).155 The onset of heart failure is uncommon (4.5%) since the liver itself prevents a significant increase in venous return. Some patients have symptoms of diarrhoea (4.5%), likely due to ischaemia caused by mesenteric vasculature steal or congestion.163 The physical examination can show a murmur in 33% of cases and, if the fistula measures more than 4mm, a thrill can be identified.164,165 In the case of liver cirrhosis, the development of an APF can go unnoticed, attributing the decompensation in question to the liver disease itself.
Congenital APFs are a cause of severe childhood PHT. They are more likely associated with heart failure since intrahepatic sinusoidal resistance is minimal, and with diarrhoea.155
DiagnosisThe initial evaluation can be carried out with Doppler ultrasound, later confirming the diagnosis with an abdominal CTA or MRA.166 These imaging techniques make it possible to visualise the dilated portal vein and artery and, using Doppler ultrasound, a turbulent portal flow that can be hepatopetal or hepatofugal.167 The fistula itself can be seen in the arterial phase, 20–30s after the injection of contrast. Angiography is useful, in addition to establishing the diagnosis, for planning treatment.155
TreatmentThis depends on the location, size and number of APFs. Small fistulas normally resolve on their own. The proposed management in these cases is monitoring via Doppler ultrasound. In cases in which the fistula increases in size or symptoms appear, treatment is recommended. The treatment of choice is embolisation, with more than one procedure being necessary on occasion.168,169 If this is not possible or fails, surgery must be considered.170 In cases of congenital APF, given its complexity, individual assessment of each case is recommended at a reference paediatric centre. The proposed treatment can be ligation, embolisation, resection or even liver transplant.159
Recommendations- •
Diagnosis of APF should be considered in patients with PHT without clear aetiology, especially if a history of an invasive liver procedure, trauma or an abdominal wound is present (B1).
- •
The diagnosis of an APF should be confirmed with an abdominal CTA or MRA (B1).
- •
Large APFs or those associated with PHT are those that require embolisation treatment. Monitoring via imaging tests is recommended for the rest of APFs (B2).
These are malformations due to disturbances in embryo development. There are few reported cases, although diagnosis is increasing due to wider use of imaging techniques. Most patients are diagnosed in childhood, although some cases are identified in adulthood. This finding should always be taken into account, given that it can involve development of complications. They are classified as intrahepatic or extrahepatic connections.171
Intrahepatic portosystemic connectionsThey are defined as connections between the portal system and systemic venous circulation, with a diameter greater than 1mm, located at least partially intrahepatically.172 Based on their morphology, four distinct types have been described (Table 4).173
Classification of the intrahepatic portosystemic collaterals proposed by Park et al.173
| Type 1 | Single large communication between the right portal vein and the inferior vena cava |
| Type 2 | Presence of one or multiple collaterals between the portal system and hepatic veins, of peripheral location limited to one liver segment |
| Type 3 | Aneurysmal connection between the portal vein and the hepatic vein |
| Type 4 | Multiple connections between the peripheral portal system and the hepatic veins, with diffuse distribution in both lobes |
Also known as Abernethy malformations, these are a group of rare malformations in which there is direct drainage from the splanchnic venous circulation to the systemic venous return, bypassing passage through the liver. In these patients, the liver is perfused by the hepatic artery in the absence of the portal vein. Venous drainage of the connection can take place in the inferior vena cava, the left renal vein, the right atrium, the iliac veins, the left hepatic vein or the azygous vein.174
These malformations are classified as type I or type II based on the complete absence of the portal vein or the presence of a vestige thereof (Fig. 2). Type I is classified into subtypes Ia and Ib (Table 5).175

Classification of congenital extrahepatic connections proposed by Morgan and Superina in 1994. Modified by Hu.174
Classification of congenital extrahepatic connections (Abernethy malformation) proposed by Morgan and Superina.175
| Type I | Absence of intrahepatic portal vein |
| Type Ia | The superior mesenteric vein and the splenic vein drain separately |
| Type Ib | The superior mesenteric vein and the splenic vein form a common trunk |
| Type I | Presence of a significant connection, with persistence of intrahepatic portal system (often hypoplastic) |
The symptoms range from asymptomatic to severe hepatic encephalopathy. Encephalopathy is due to the presence of the connection, not to liver dysfunction. An association has been suggested between the worsening of symptoms and age, resulting in some patients being diagnosed in adulthood.176,177 In most cases, liver function is conserved and the absence of manifestations of PHT is characteristic (splenomegaly, oesophageal varices or ascites). Over the course of the disease, appearance of nodular liver lesions (focal nodular hyperplasia, nodular regenerative hyperplasia, adenoma, hepatoblastoma and even hepatocellular carcinoma) is common.178–180
There are associated congenital malformations, such as biliary atresia and heart, genitourinary or musculoskeletal system malformations. At the heart level, symptoms are aggravated by the increase in venous return through the connection. The most commonly-identified complications are patent foramen ovale, defects in the atrial or ventricular septum, patent ductus arteriosus, coarctation of the aorta, dextrocardia, etc.172,174 In the respiratory system, despite the absence of PHT, the onset of hepatopulmonary syndrome has been described, such as portopulmonary hypertension due to the passage of substances through the connection. In both cases the patient had dyspnoea, with the addition of orthodeoxia in hepatopulmonary syndrome.181,182
DiagnosisThe diagnosis should be established via abdominal CTA or MRA.183 Doppler ultrasound is especially useful in prenatal screening. In addition to observing the portosystemic venous connection, it can show compensatory hypertrophy in the hepatic artery. It is important not to confuse the absence of a portal vein with PVT.174 These imaging techniques can also help complete the patient's assessment, allowing for diagnosis of other malformations and an examination of the hepatic parenchyma. Angiography is useful both for establishing the diagnosis and for planning treatment.
TreatmentThere is limited experience in the management of these malformations.172 In general, monitoring is recommended in asymptomatic cases. For symptomatic cases, the treatment options depend on the malformation type. Some small intrahepatic connections detected in the first months of life have resolved spontaneously.184,185 Therefore, monitoring of evolution is recommended until age 2. If the connection does not resolve and the patient has symptoms, embolisation or surgical resection should be considered.186,187
In type I Abernethy malformations there is no surgical technique that can reconstruct the portal vein. Consequently, the available options are liver transplant or, in the case of localised tumours, resection thereof.174 In type II Abernethy malformations, when medical management is not sufficient, the most recommended alternative is embolisation of the connection. Before the procedure it is important to make sure that the portal system exists and receives adequate blood flow, or the resulting complications can be serious and involve the consequent development of PHT.188
Recommendations- •
The presence of a congenital portosystemic connection should be suspected in cases of hepatic encephalopathy not explained by another cause (A1).
- •
The diagnosis of congenital portosystemic connections should be confirmed with an abdominal CTA or MRA (B1).
- •
In patients with congenital intrahepatic connections (diagnosed at birth) that do not resolve by age 2 and are symptomatic, embolisation or surgical treatment should be considered, making sure that the liver receives adequate portal blood flow (B2).
- •
The recommended treatment in symptomatic cases of type I Abernethy malformation is liver transplant (B2).
- •
Embolisation or ligation of type II Abernethy malformations is recommended as long as it is symptomatic, making sure that the liver receives adequate portal blood flow (B1).
- •
In Abernethy malformations, it is recommended to evaluate the association with other malformations and to specifically monitor to detect the onset of lung complications and liver tumours (B2).
The finding of portosystemic collaterals is common in cirrhotic patients with hepatic encephalopathy clinically shown to be refractory to treatment (defined as recurrent or persistent encephalopathy). Depending on the series, the percentage is variable, ranging from 46 to 70%.189–191
Initial management is similar to the rest of patients with hepatic encephalopathy: as a first step, it is recommended to identify and correct the possible precipitating factor, administer non-absorbable disaccharides to achieve 1–2 bowel movements per day, and, as a second step, introduce non-absorbable antibiotics such as rifaximin. In the event of relapse or absence of response, an alternative proposed treatment is embolisation of the large collaterals, a procedure carried out using interventional radiology techniques. The materials used (coils, amplatzer, matrix) and the access routes (femoral, transjugular, transparietohepatic) vary based on the case and the centre's experience.
In the latest European and American encephalopathy management guidelines,192,193 a therapeutic option in refractory cases is embolisation of large collaterals in patients with good liver function. According to a retrospective multicentre study that analysed the experience in the largest published series to date (37 patients), the cut-off point that allows for prediction of a good response is liver function with a score less than or equal to 11 points on the MELD scale. Regarding complications, in addition to the risks inherent in the technique, the increased portal pressure arising from the embolisation can foster the onset of ascites or haemorrhage. Nevertheless, in well-selected patients (without oesophageal varices or with small varices and without ascites or with mild, well-controlled ascites) the procedure has been shown to be safe, with no evidence of a significant increase in complications.194,195
Recommendations- •
In cirrhotic patients with recurrent or persistent hepatic encephalopathy refractory to medical treatment, the presence of large portosystemic collaterals should be investigated via CTA and/or MRA (A1). In these cases embolisation of the dominant collateral is recommended, especially in patients with good liver function (B1).
Hereditary haemorrhagic telangiectasia (HHT) or Osler–Weber–Rendu disease is a genetic disease of autosomal dominant inheritance, characterised by the appearance of abnormal vascular structures (telangiectasia or arteriovenous malformations) that can appear in any organ, being responsible for the associated morbidity and mortality. The penetrance of symptoms is variable and generally increases with age. It is 90% at 45 years of age.196 Epistaxis is the most common symptom (up to 96% of patients may have nose bleeds) and is generally the first to appear.197 HHT is included within rare diseases since its mean prevalence is estimated around 1:5000–1:8000,198 although isolated populations exist where the rate is higher due to founder effect (Jura in France, Funen Island in Denmark and the Dutch Antilles).199 It is still an underdiagnosed condition200 and has no definitive cure.
The diagnosis is based either on the Curaçao Criteria201 (Table 6): epistaxis, telangiectasias, familial clustering of a dominant aspect and internal organ involvement (with at least three of the above being necessary to confirm the diagnosis),202,203 or on the identification of the causal mutation by conducting a molecular study. New techniques to facilitate diagnosis, such as infrared spectroscopy, seem to be useful in identifying patients.
Curaçao clinical criteria for the diagnosis of hereditary haemorrhagic telangiectasia.
| Epistaxis | Spontaneous and recurrent |
| Telangiectasia | Multiple and in characteristic locations: lips, oral cavity, fingers, nose |
| Familial clustering | Family history or first-degree relative with HHT (the affected relatives should present at least two of the highlighted symptoms) |
| Involvement of internal organs | Arteriovenous malformations: pulmonary, hepatic, cerebral or spinal Telangiectasia in the gastrointestinal tract |
Uncertain diagnosis: 1 criterion. Probable diagnosis: 2 criteria. Certain diagnosis: 3 or 4 criteria.
To date, three genes involved in the development of the disease have been identified (Table 7). The first one described was the ENG gene, located in chromosome 9 (9q33-q34) which encodes the endoglin protein; mutations in this gene give rise to HHT type 1.204–206 In HHT type 2, the mutation is found in the ACVRL1 gene which encodes the activin receptor-like kinase 1 (ALK1) protein, specifically the 12q11-q14 region of chromosome 12.207,208 Some 2% of patients have a combined HHT and hereditary juvenile polyposis syndrome. In this case, the mutation is present in the MADH4 gene, located in chromosome 18.209 This group of patients has phenotypical features of HHT and high risk of malignant tumours in the gastrointestinal tract at early ages.210
Genes involved in hereditary haemorrhagic telangiectasia.
| Type | OMIM number | Chromosome | Involved gene | Protein |
|---|---|---|---|---|
| HHT 1 | #187300 | 9 | ENG | Endoglin |
| HHT 2 | #600376 | 12 | ACVRL1 | ALK1 |
| JPHT | #175050 | 18 | MADH4 | Smad 4 |
| HHT 3 | 5 | Not identified | ||
| HHT 4 | 7 | Not identified | ||
| CM-AVM | 14 | BMP9 (GDF2) | BMP9 |
OMIM: Online Mendelian Inheritance in Man; HHT: hereditary haemorrhagic telangiectasia; JPHT: juvenile polyposis hereditary telangiectasia; CM-AVM: capillary malformation-arteriovenous malformation syndrome.
Mapping techniques have identified at least two other loci, one located in chromosome 5 and the other in chromosome 7,211,212 potentially involved in the disease genesis, although the causative genes have still not been identified. Recently, a new syndrome has been reported (capillary malformation-arteriovenous malformation syndrome) with a phenotype similar to that of HHT, caused by mutations in the gene that encodes the bone morphogenetic protein 9 (BMP9).213
All of these proteins have in common being involved in the triggering of extracellular–intracellular signalling of the TGF-β superfamily, involved in multiple biological effects such as regulation of cellular proliferation, differentiation, migration and formation of the extracellular matrix, maintenance of tissue homoeostasis and repair in adults. There seems to be geographic variability with the prevalence of HHT type 1 in North America, English-speaking countries and Northern Europe, while HHT type 2 is more common in the Mediterranean region and South America.198,214,215 Considering the genotype-phenotype relationship, patients with HHT type 1 seem to have more risk of presenting vascular malformations at the pulmonary level and in the central nervous system, while patients with HHT type 2 have higher rates of liver and gastrointestinal tract involvement.216,217
SymptomsHepatic involvement in HHT can present as telangiectasias, confluent vascular masses, hepatic artery dilatation and short circuits of various types (arterioportal, arteriovenous and/or venoportal).218,219 Based on the different series, liver involvement appears in 30–84% of patients with HHT.220–222 It is more common in patients with HHT type 2 and in women.199 The increased diameter of the common hepatic artery seems to be one of the earliest signs of liver involvement223 and it is also one of the most specific findings of liver disease in patients with HHT.224,225
In cases of severely compromised liver (8%)226 complications secondary to the development of different types of arteriovenous or venoportal connections can develop which, according to the classification created by García-Tsao et al.,218 can be:
- •
Type 1. Short circuit between the hepatic artery and the suprahepatic veins with high-output heart failure secondary to hyperdynamic circulation caused by excess flow. It is the most common (63%) of the complications secondary to large hepatic fistulas227 and progresses with dyspnoea on exertion, orthopnoea, peripheral oedema, oliguria and ascites.
- •
Type 2. Short circuit between the hepatic artery and portal vein (17% of cases), with hepatic pseudocirrhosis (due to the increase in sinusoidal flow with fibrous tissue deposit) and PHT228 or PHT secondary to nodular regenerative hyperplasia.229 The accompanying symptoms are hydropic decompensation, anaemia, risk of bleeding due to congestive gastropathy or oesophageal varices, splenomegaly and hepatic encephalopathy.230,231
- •
Type 3. Biliary ischaemia (19% of cases) secondary to vascular steal (biliary vascularisation originates in the hepatic artery). In the case of large arteriohepatic or arterioportal short-circuiting, biliary ischaemia can occur with stenosis of the bile ducts and biliary necrosis with the formation of bilomas, cholestasis and, occasionally, cholangitis. The symptoms correspond to the classic triad of pain in the right hypochondrium, occasionally with fever and jaundice.232
Another type of fistula is portal-hepatic (between the portal vein and suprahepatic veins) with potential risk of developing nodular regenerative hyperplasia. Another less common symptom is mesenteric ischaemia due to vascular steal in arteriohepatic short-circuits.233
García-Tsao et al. observed that the mean age for the diagnosis of high-output heart failure, PHT and bile duct involvement in HHT was 52, 62 and 39 years, respectively; figures that coincide with later studies.234
In a long-term follow-up study (mean of 44 months) of patients with HHT and liver involvement, a 5.2% mortality was observed related to liver disease (mortality rate of 1.1% and morbidity of 3.6% for every 100 patients/year) with an effective response to treatments administered (medical, surgical and interventional radiology) of 63%.235
Diagnosis and screeningThe presence of diffuse vascular hepatic malformations should orient towards the diagnosis of HHT, although there are other syndromes with an even lesser prevalence, such as Klippel-Trénaunay, that can also present these types of changes.41
Although there is contention about the convenience of screening for liver involvement due to the relatively low percentage of associated symptoms (8% of patients have heart failure, PHT, encephalopathy or cholangitis),233,236 recent prospective studies in patients with HHT showed substantial rates of morbidity and mortality (1.1% and 3.6% per year, respectively), which recommends both the conduct of screening for possible liver involvement and the establishment of a monitoring protocol.234 Additional reasons to proceed with liver/abdominal studies are the support that it provides for the diagnosis of the disease based on the Curaçao Criteria and the fact that, in some studies, pharmacokinetic changes have been observed in patients with liver involvement,237 as well as a greater predisposition to the development of heart complications in patients with focal nodular hyperplasia and an increase in the hepatic artery diameter.224
For the diagnosis of liver involvement in HHT, imaging studies based on abdominal ultrasound and computed tomography (CT) are necessary. At the ultrasound level, it is important to observe the size of the hepatic artery (usually dilated) and to determine the peak systolic velocity therein220,238; also, a change in the portal flow and the suprahepatic veins on the Doppler allows for diagnosis of the presence of arterioportal and arteriohepatic short circuits.239 Ultrasound studies allow for stratification of the severity of hepatic lesions in HHT by using the classification proposed by Buscarini et al.220 (Table 8), in which asymptomatic patients and those with degree 0, 1 and 2 involvement do not require treatment, while for those with degrees 3 and 4 additional studies would be recommended (echocardiogram with estimated calculation of cardiac output and lung pressure and endoscopic studies, respectively, to identify the presence of high-output heart failure or PHT).
Degrees of severity of hepatic vascular malformations in patients with hereditary haemorrhagic telangiectasia according to Doppler ultrasound.
| Degrees | Parameters |
|---|---|
| Degree 0 | Hepatic artery diameter>5<6mm and/or |
| Peak systolic velocity of the hepatic artery>80cm/s and/or | |
| Hepatic arterial resistive index<0.55 and/or | |
| Peripheral hepatic hypervascularisation | |
| Degree 1 | Extrahepatic dilatation of hepatic artery>6mm and |
| Peak systolic velocity of the hepatic artery>80cm/s and/or | |
| Hepatic arterial resistive index<0.55 | |
| Degree 2 | Dilatation of the extra- and intrahepatic hepatic artery (two channels) and |
| Peak systolic velocity of the hepatic artery>80cm/s | |
| Moderate changes in portal and/or hepatic vein flow | |
| Degree 3 | Tortuous hepatic arteries and/or |
| Moderate dilatation of the portal and/or hepatic veins | |
| Degree 4 | Marked dilatation of the portal and/or hepatic veins |
| Marked abnormalities in the portal and splenic flow in the Doppler study |
Abdominal CTA seems to be more sensitive than Doppler ultrasound both for the detection of small vascular lesions and for the indirect diagnosis of fistulas and large hepatic arteriovenous malformations.222,240 Therefore, and despite the associated radiation (to be considered especially in paediatric patients), it would be advisable to assess its conduct at least at the time of diagnosis, which would also help with the diagnosis of vascular malformations in other abdominal organs such as the pancreas, where arteriovenous malformations are observed in 33–42% of cases (more common in patients with HHT 2),241–243 spleen, gastrointestinal tract wall and kidneys. Another common finding in these patients is the existence of anatomical variants of normality mainly at the origin of the hepatic artery and its branches in the coeliac trunk and superior mesenteric artery.244
Magnetic resonance seems to have the same sensitivity as CT for diagnosing liver involvement in HHT with the added advantage of the absence of ionising radiation, although the series of patients and experience with this technique are currently much lower.245
Abdominal arteriography is usually reserved for cases in which a haemodynamic or pre-transplant study is planned.222
Since the majority of patients with HHT receive a routine echocardiogram with contrast (agitated saline solution) to screen for right-left short-circuiting to diagnose pulmonary arteriovenous malformations, it would be recommended to simultaneously estimate cardiac output and lung pressures. Endoscopic studies will be reserved for patients with active bleeding, anaemia not justified by the degree of nose bleeds and for the screening and treatment of oesophageal varices and PHT gastropathy, if applicable.
Biochemically, the presence of anicteric cholestasis is observed in up to one third of patients and seems to be correlated with the extent of the vascular malformations.235 Systems to evaluate the severity of liver involvement in patients with HHT are being developed, based on logistic regression models that consider, in addition to alkaline phosphatase and haemoglobin levels, radiological involvement in ultrasound or CT and the age and sex.234 The vascular changes observed in patients with HHT foster the development of foci of focal nodular hyperplasia, a finding observed in 16% of cases.234
The conduct of liver biopsies in patients with HHT should be avoided if possible due to the high risk of haemorrhagic complications.41
Treatment1.1.15General measuresCurrently, there are no treatment indications in asymptomatic patients with or without liver involvement.
For symptomatic cases, the treatment options depend on the type of associated complication. In the most common extrahepatic complication, high-output heart failure, intensive treatment is indicated in NYHA functional classes II to IV, based on the general corrective measures for anaemia: low-sodium diet, diuretics, beta blockers, digoxin, ACE inhibitors, cardioversion or radiofrequency ablation when there are associated rhythm disorders. The objective would be to alleviate symptoms and stabilise the haemodynamic situation.203,246
Regarding the complications of PHT (bleeding due to oesophageal varices, ascites, encephalopathy), the treatment should be the same as that for patients with PHT with other aetiologies.219 Adequate anaemia support treatment (iron therapy and/or blood transfusions) is vital in this group of patients.
1.1.16Pharmacological treatmentApproximately 63% of patients have a complete response to medical treatment, while 21% have a partial response. The response to treatment should be evaluated 6–12 months after it is started. In the case of cardiac overload, ACE inhibitors and carvedilol can be used to prevent cardiac remodelling. The latter, along with the usual non-cardioselective beta blockers (nadolol and propranolol), can be used to prevent haemorrhage due to PHT in patients with clinically-significant oesophageal varices or with gastrointestinal telangiectasias.235 Also, propranolol, due to its apoptotic and anti-angiogenic effect, seems to be able to decrease nose bleeds.247 The invasive management detailed below is intended for patients who do not respond to medical treatment.
Bevacizumab, an anti-angiogenic monoclonal antibody, has been shown, in a single study with 24 patients with large hepatic vascular malformations and high cardiac output, to be able to improve cardiac output and shorten the duration of epistaxis episodes (complete response 12% and partial response 70%, respectively). However, these data should be interpreted cautiously since it has a non-negligible toxicity (hypertensive crisis, proteinuria, risk of thromboembolic complications) and causes changes in the angiogenesis process that could foster anastomotic dehiscence or infection of the surgical wound in the case of emergency liver transplant.248
1.1.17Endoscopic treatmentIn the case of accessible bleeding gastrointestinal telangiectasias, endoscopic treatment with argon gas is the treatment of choice, given its ease of use, efficacy and safety. Concomitant pharmacological treatment with endoglin production stimulators or antiangiogenics, such as bevacizumab, is not currently recommended.249–252
1.1.18Endovascular treatmentEndovascular treatment via transarterial embolisation of vascular malformations should only be used in patients with cardiac involvement, incapacitating symptoms, without response to medical treatment and who are not candidates for a liver transplant, since the post-procedural mortality reaches 10%, with a non-negligible rate of complications of 20% (iatrogenic pulmonary embolisation, development of biliary abscesses, extensive parenchymal and bile duct necrosis, cholangitis, haemobilia, biliary sepsis or liver failure).253 The risk of the procedure and the reappearance of fistulas in other areas has made the liver transplant the treatment of choice today, especially when patients develop secondary cardiac involvement.
The use of TIPS in managing PHT complications is not recommended due to the high morbidity and worsening of the hyperdynamic state.219
1.1.19Liver transplantA liver transplant is the only curative alternative for hepatic venous malformations in the context of HHT, although there are reported cases of abnormal revascularisation over a graft.254
The indications for transplant, in addition to the usual ones, are ischaemic bile duct necrosis and pharmacologically-untreatable secondary cardiac involvement. After transplant, the secondary hyperdynamic situation disappears.255 As an aspect to consider, the pre-transplant evaluation should always include a right cardiac catheterisation in order to rule out severe pulmonary hypertension.
In the published series, the liver transplant is of greater technical difficulty due to the risk of arterial bleeding secondary to the hyperperfusion of the liver. Therefore, the initial ligation of the hepatic artery is essential to decrease hyperdynamic circulation and for rapid improvement in heart function. In patients with large pre-existing arterial aneurysms, complete resection of the hepatic artery will be necessary to prevent the onset of post-operative pseudoaneurysm thrombosis.
Post-operative mortality in liver transplants is 7–10%, with an excellent long-term survival rate (82–92% in a mean follow-up of 10 years).256–258
Recommendations- •
In the presence of multiple hepatic arteriovenous malformations considering a diagnosis of HHT is recommended using clinical criteria and/or genetic testing (A2).
- •
Performing a Doppler ultrasound as the first-choice technique for the diagnosis and staging of liver malformations is recommended. CTA can be considered as an alternative in cases where an experienced sonographer is not available (A1).
- •
In patients with liver involvement, especially if it is severe, a cardiology evaluation and assessment of the signs of PHT is recommended at diagnosis and during follow-up.
- •
A liver biopsy is not necessary for diagnosis. If it is planned for other reasons, assessing the risk-benefit ratio is recommended due to the high risk of haemorrhage (A1).
- •
Management of patients with HHT should be carried out at specialised centres, preferably with liver transplant units (A1).
- •
A liver transplant is the only curative option for HHT with high-output cardiac involvement, advanced liver disease or ischaemic bile duct necrosis (B1).
- •
Transarterial embolisation is a palliative, temporary and high-risk method that should only be considered in specialised centres for patients who are not candidates for a liver transplant (B2).
Sinusoidal obstruction syndrome (SOS) is secondary to obstruction of liver venous drainage that is caused by damage to the liver sinusoidal endothelium by intermediate metabolites of different toxic substances. Initially, it was called hepatic veno-occlusive disease, since sinusoidal congestion, centrilobular haemorrhagic necrosis and perisinusoidal and endovascular fibrosis were described as characteristic findings. Currently, thanks to the development of a pyrrolizidine alkaloid-induced animal model, we know that the initial phenomenon in SOS is lesion in the hepatic endothelial cells, which detach from the wall, embolise to the central area of the hepatic lobe, create a flow obstruction, and, therefore, sinusoidal congestion. Other changes also occur such as extravasation of erythrocytes and accumulation of cellular and non-cellular waste in the space of Disse, which also obstructs the blood flow through the sinusoids and makes the name SOS more appropriate.259
In our area, SOS appears in four circumstances: (1) as a complication of myeloablative regimens in haematopoietic cell transplantation (HCT); (2) after treatment with chemotherapy for solid tumours (generally metastatic colorectal carcinoma with oxaliplatin); (3) after immunosuppressant treatment such as azathioprine, 6-mercaptopurine or gemtuzumab, and (4) as a complication of radiotherapy, administered as total body or locally (radioembolisation of liver tumours). Nevertheless, there is a long list of drugs and toxic agents related to SOS (Table 9).41
Drugs and toxic agents related to sinusoidal obstruction syndrome.
| Dactinomycin |
| Azathioprine |
| Busulfan |
| Carmustine |
| Cytosine arabinoside |
| Cyclophosphamide |
| Dacarbazine |
| Gemtuzumab |
| Mercaptopurine |
| Mitomycin |
| Oxaliplatin |
| Pyrrolizidine alkaloids |
| Urethane |
| Terbinafine |
| Traditional eastern medicinal herbs |
| 6-Mercaptopurine |
| 6-Thioguanine |
| Total body and local radiotherapy |
| Transfusion of platelets containing ABO-incompatible plasma |
The wide variability in the symptoms of SOS and the heterogeneity of its diagnostic criteria make it difficult to establish its real incidence. In the West, the rate of SOS after HCT is between 5 and 50%.1 This variability is due to the fact that the risk of SOS differs based on the type of myeloablative treatment, the number of chemotherapy cycles and patient-specific factors. SOS is most common in patients treated with cyclophosphamide or busulfan, in those who have received more than four chemotherapy cycles and in those with pre-existing liver disease. Identifying risk factors has allowed for treatment to be optimised and the incidence to be reduced to around 5%.260 The highest incidence of SOS in patients receiving HCT may be due to the fact that the treatment simultaneously damages both sinusoidal liver cells and haematopoietic stem cells, which prevents them from replacing the damaged sinusoidal cells.261
In recent years, histologic changes specific to SOS have been reported in liver tissues from the resection of hepatic colorectal carcinoma metastasis in patients who had received treatment with oxaliplatin. These changes, and nodular regenerative hyperplasia, are observed in 35–75% of these patients.262,263 The addition of bevacizumab, an anti-angiogenic drug, seems to have a protective effect.264 In the pathogenesis of these findings, an inflammatory mechanism with activation of the vascular endothelial growth factor (VEGF) was invoked, secondary to chemotherapy-induced tumour necrosis.
Azathioprine and 6-mercaptopurine have been traditionally associated with the development of SOS, but the mechanism by which they induce sinusoidal damage is unknown. Gemtuzumab is an antibody whose primary target is the myeloid cells that express CD34, an antigen that they share with liver sinusoidal cells. This monoclonal antibody was used for the treatment of acute myeloid leukaemia, along with other potent cytotoxic agents, between 2000 and 2010, causing SOS in up to 9% of patients, with a mortality of 2.7%.265
SymptomsThere are three forms of SOS symptoms: (1) acute, characterised by rapidly-progressing ascites, painful hepatomegaly and jaundice; (2) subacute, with recurring ascites, splenomegaly and hepatomegaly, and (3) chronic, indistinguishable from the symptoms of PHT associated with cirrhosis, either asymptomatic or manifesting as complications of PHT.
In patients receiving HCT, SOS tends to appear in the first 20 days after treatment with cyclophosphamide, although it can take longer than a month if other less aggressive treatments have been used. It is characterised by hyperbilirubinaemia, difficult-to-control ascites and painful hepatomegaly. These findings, although characteristic of SOS, can follow other aetiological factors, common in the context of an HCT, such as acute viral hepatitis, bacterial infections, hepatotoxicity or graft versus host disease. This makes the application of purely clinical diagnostic criteria, such as the Seattle or Baltimore criteria, insufficient for the diagnosis of SOS after HCT.266,267
SOS is a severe complication of HCT, with mortality rates that vary between 3% in mild forms to 100% in severe cases. The degree of hyperbilirubinaemia and hypertransaminasaemia, weight gain and increased hepatic venous pressure gradient are associated with a worse prognosis.
SOS associated with oxaliplatin chemotherapy tends to be asymptomatic or mildly symptomatic in its acute form and tends to manifest later with unequivocal signs of PHT (oesophageal varices, thrombocytopenia or splenomegaly). The correlation between the histological and clinical findings is minimal, since the majority of the liver tissue samples analysed come from resection material from hepatic metastases that are only carried out in cases of conserved liver function. The impact of SOS on the prognosis for these patients is uncertain, and it has not been shown that its presence increases mortality. The SOS regression capacity after suspension of oxaliplatin chemotherapy is unknown, and it seems that between 1 and 3 years should pass in order to observe an improvement in indirect parameters of PHT, such as spleen diameter.
DiagnosisIn our area, SOS should be suspected in two very different clinical contexts: immediately after an HCT and after extended treatment with chemotherapy for colorectal carcinoma or with some immunosuppressants.
Regarding the first of the situations, the suspicion of SOS is based mainly on the onset of jaundice, hepatomegaly and weight gain or ascites after an HCT. The diagnostic criteria published in the 1980s were solely based on clinical findings, and, when they are present, the disease is already at a very progressed stage.266,267 For this reason, the European Group for Blood and Marrow Transplantation has established new criteria that combine clinical, histological and imaging findings and has described the chronology of their onset (Table 10).268
Diagnostic criteria of sinusoidal obstruction syndrome (SOS) after haematopoietic cell transplantation (HCT)268
| Classic SOS (in the 21 days after HCT) | Late-onset SOS (>21 days after HCT) |
|---|---|
| Bilirubin≥2mg/dl and two of the following: | Classic criteria or |
| Painful hepatomegaly | Histological confirmation or |
| Weight gain>5% | Two of the following: |
| Ascites | Bilirubin≥2mg/dl |
| Painful hepatomegaly | |
| Weight gain>5% | |
| Ascites | |
| and haemodynamic or ultrasound evidence of SOS |
Analytically, elevated bilirubin is a sensitive but non-specific index. Although in the past the possible diagnostic and prognostic usefulness of various haemostatic factors such as proteins C and S, antithrombin, factor VIII or fibrinogen, and some endothelial damage markers, such as thrombomodulin or the von Willebrand factor were analysed,269,270 currently they lack practical applicability.
Imaging tests are vital, since they can rule out other diseases such as Budd–Chiari syndrome or acute portal vein thrombosis, and they offer some findings that, although non-specific, can support the diagnosis. The most common ultrasound signs in Doppler studies are hepatomegaly, splenomegaly, ascites, thickening of the vesicular wall, increased portal calibre with flow inversion and loss of the typical triphasic flow pattern in hepatic veins.271
Liver biopsy is important, since it makes it possible to establish the diagnosis and exclude other conditions with similar symptoms after an HCT. It should be performed transjugularly to minimise the risk of complications and measure the hepatic venous pressure gradient. A hepatic venous pressure gradient above 10mmHg has been linked to a worse prognosis and is closely correlated with the findings of a liver biopsy, with a specificity of 91% and a positive predictive value of 86%.272 The most characteristic histopathological findings are congestive sinusoidal obstruction, which can be associated with perisinusoidal fibrosis, fibrotic central venous obstruction, nodular regenerative hyperplasia and hepatic peliosis.273
Chemotherapy-induced SOS for the treatment of colorectal carcinoma should be suspected in patients with unequivocal signs of PHT (mainly the presence of oesophagastric varices, thrombocytopenia or splenomegaly), in the absence of clinical, radiological or elastographic data suggestive of cirrhosis. In these patients, it is essential to conduct a full liver disease aetiological study that includes liver biopsy. The biopsy will show findings similar to those described in the context of an HCT, to which chronic changes, such as nodular regenerative hyperplasia, can be added. Doppler ultrasound is required to check the permeability of the large hepatic veins and to evaluate the presence of signs of cirrhosis, such as irregular liver contour. The development of new nuclear magnetic resonance imaging techniques will facilitate the radiological diagnosis of SOS.274,275 SOS attributed to immunosuppressants, such as azathioprine, is usually suspected in progressive thrombocytopenia and maintained in patients who receive said treatments and, just like in chemotherapy-induced SOS, it must be histologically confirmed.
Prophylaxis and treatmentIt is essential to try to minimise the risk and severity of SOS, since no completely effective treatment is available. Prophylactic measures include acting on modifiable risk factors, such as reducing the intensity of conditioning in HCT or avoiding concomitant use of hepatotoxic drugs with potentially-SOS-causative agents. Furthermore, patients with a higher risk of developing the disease should be identified in order to use non-ablative regimens and monitor them closely during treatment.271,276
There is also the option of using pharmacological prophylaxis. To this end, a meta-analysis has analysed the usefulness of anticoagulants, with a global negative result.277 A prospective study showed that patients who received ursodeoxycholic acid after an HCT had less severe SOS and a one-year survival rate higher than those who did not receive it.278 However, a recent review of the four published studies that compare ursodeoxycholic acid with placebo or no treatment concluded that the evidence supporting the prophylactic use of ursodeoxycholic acid is of low quality.279
On the other hand, a multicentre controlled study showed that defibrotide, an oligodeoxyribonucleotide with antithrombotic and anti-inflammatory properties, decreases the incidence of SOS after HCT in patients under 18 with at least one risk factor for developing SOS. In adults, only retrospective studies have been conducted, but there are no reasons that impede extrapolation of the results obtained in children.280
Treatment for SOS is mainly supportive, with adequate management of water retention and monitoring for possible complications. The most serious cases should be managed in an intensive care unit.281 TIPS has been used in some patients with varying results.282,283 The indication for liver transplant is limited by the frequently oncological nature of the underlying disease, although it has been performed successfully in select patients.284 The only drug that has shown efficacy for the treatment of severe SOS is defibrotide, which demonstrated an increase in survival in patients with SOS and multiple organ failure at a dose of 25mg/kg/day compared to historical controls (38.2% vs 25%; p=0.0109).285
Recommendations- •
Consider a diagnosis of SOS in the event of signs suggestive of liver disease or PHT in patients who have received a bone marrow transplant, chemotherapy or treatment with immunosuppressants due to a solid organ transplant or to control an inflammatory bowel disease (B1).
- •
Consider a diagnosis of SOS in the presence of hepatomegaly, hyperbilirubinaemia, weight gain and ascites in patients who have undergone a bone marrow transplant and/or are undergoing cytostatic or immunosuppressant treatment (B1).
- •
A diagnosis of SOS is established by the data provided by the imaging techniques and, especially, the haemodynamic study and transjugular liver biopsy (B1).
- •
Prophylaxis for SOS is based on controlling risk factors and treatment as a support measure (B1).
- •
Defibrotide is effective as prophylaxis in patients with risk factors and as treatment for SOS cases with multiple organ failure (B1).
- •
There is no evidence to recommend the use of TIPS in patients in whom medical treatment fails.
Imaging techniques are gaining increasing importance in the diagnosis and evaluation of vascular liver diseases, not just as a non-invasive tool to establish a diagnosis, but also to evaluate the degree of involvement, monitor response to treatment and detect associated complications.
Doppler ultrasound continues to be the basic tool and it is, without a doubt, the first test to be performed in any vascular liver disease.1,286,287 It is a harmless, non-invasive technique with high availability. The sensitivity of Doppler ultrasound to detect flow in blood vessels, even those of small calibre, is very high. The findings do not generally require confirmation via other imaging tests.288 It has the added advantage of allowing for a functional study, offering information about the direction and velocity of the flow. Contrast ultrasound can be helpful on occasion, since it increases the sensitivity of the baseline ultrasound for flow detection.289–291
Elastography allows for data to be collected about the physical properties of the liver and spleen tissue by measuring the velocity of shear waves that are generated through the transmission of mechanical or ultrasound impulses in the tissues.292 Since there is a notable fibrotic component in liver tissue in cirrhosis, the rigidity of the liver is high in this disease292; however, high degrees of liver fibrosis are generally absent in vascular liver diseases and hepatic elastography in the latter shows normal or only slightly elevated results. Therefore, hepatic elastography is an additional tool that can be used for a correct differential diagnosis.
CT and MRI, both with intravenous contrast, can be useful to reach a diagnosis when the ultrasound image is not good.293,294 They also allow for assessment of the permeability of the full splenoportal-mesenteric axis, many times partially hidden in ultrasound due to the interposition of abdominal gas,87,295 and they offer a map of collateral circulation of the entire abdomen, which is also impossible to obtain in its entirety via ultrasound.296–301 The images obtained with CT and MRI have the great advantage of being suitable for evaluation in multidisciplinary committees for therapeutic planning.1 There are insufficient comparative studies that provide data on the diagnostic superiority of one of the two techniques over the other in the evaluation of liver vessels. The advantages of CT over MRI are greater availability, lower cost and it is faster to carry out, leading to it being better tolerated by patients and it is subject to fewer movement artefacts. However, the great disadvantage is the use of ionising radiation, which limits its use when repeated examinations are required.302 In the case of allergies to CT iodinated contrast, the alternative is to perform an MRI. However, both the iodinated contrast used in CT and the gadolinium contrast for MRI are contraindicated if there is kidney failure, the first one because it is nephrotoxic, and the second one because it has been associated with nephrogenic systemic fibrosis.303 MRI studies without contrast with specific sequences have been proposed to be able to study venous abdominal vessels,304,305 although studies with contrast are always preferable.
Radiology in Budd–Chiari syndromeUltrasound, CT and MRI are very sensitive when evaluating the permeability of suprahepatic veins and the inferior vena cava, with a precision of >95%.306 Therefore, ultrasound is the initial technique for evaluating a patient with clinical suspicion of Budd–Chiari syndrome (BCS).1,41 It is recommended that in patients with suspected BCS, the ultrasound be performed in centres with extensive experience.
1.1.20Direct signsThe diagnosis of BCS is confirmed by directly observing involvement of one or more suprahepatic veins.307 This can manifest in different ways: (1) echogenic material (thrombus) inside one or more suprahepatic veins, with the absence of flow in the entire vessel or distally to the thrombosis (in acute or sub-acute cases); (2) total absence of one or more suprahepatic veins, replaced by a hyperechogenic cord (fibrosis), with the absence of suprahepatic flow confirmed by colour Doppler and pulsed-wave Doppler, and (3) replacement of the hepatic vein by a tortuous vessel of abnormal morphology.
Although in our area inferior vena cava involvement is not common (10–15%), in all patients with suspected or confirmed diagnosis of BCS, Doppler ultrasound should explore the permeability of the inferior vena cava.307
In the vast majority of cases, the B-mode examination, colour Doppler and pulsed-wave Doppler is sufficient to rule out or diagnose BCS. In complex cases in which there is still uncertainty about the presence of thrombosis in one or more hepatic veins after colour/weighted/pulsed-wave Doppler, the use of ultrasound contrast can be of great help to confirm or rule out this diagnosis.
Despite the fact that the three techniques (ultrasound, CT and MRI) allow for detection of thrombi inside the suprahepatic veins with similar precision, ultrasound seems superior in the detection of membranes in the inferior vena cava and of short stenosis in the suprahepatic veins.308
1.1.21Indirect signsHepatic morphologyIn acute cases, nearly invariably the liver is greatly increased in size and is very heterogeneous, sometimes with the presence of nodules (see below).309,310 The liver surface tends to be smooth and ascites tend to be present. CT and MRI can show disturbed perfusion of the hepatic parenchyma, something which cannot be confirmed with ultrasound.309–311 The more peripheral liver regions are more congested and oedematous. Therefore, contrast studies are characterised by increased uptake in the more central areas of the liver and decreased uptake in the peripheral parenchyma. The areas less involved in drainage, such as the caudate lobe, show a relative increase in contrast uptake during the venous phase.309–313 On the MRI, peripheral oedema shows as a hypointense signal on T1-weighted sequences and a hyperintense signal on T2-weighted sequences in the more peripheral areas.
In chronic phases, an aspect of the liver similar to a cirrhotic liver can be observed, with atrophy of the segments that have insufficient venous return depending on the involved vein(s) (e.g., atrophic right lobe).309–311 In CT/MRI, the central uptake pattern is lost, occasionally appearing with heterogeneous uptake without a defined pattern or showing homogeneous uptake. Also, decreased oedema in chronic forms causes a decrease in the hyperintense signal on T2-weighted sequences.311,314 In 80% of sub-acute or chronic cases, the caudate lobe is hypertrophic (see below). Hypertrophy of the caudate lobe can be very pronounced and lead to compression of the inferior vena cava in its intrahepatic section.307,309–311
In patients with untreated BCS, hepatic elastography shows a marked increase in liver rigidity due to venous congestion (unpublished data); therefore, liver rigidity should not be used to detect the presence of cirrhosis in these patients.
Caudate veinThe caudate lobe has venous drainage independent of the inferior vena cava in the form of a single caudate vein (85–90%). This vein is usually not affected by the thrombosis and, in the case of thrombosis of the other veins, hepatic venous drainage occurs through it, leading to hypertrophy of the vein and the caudate lobe. In 50% of patients with BCS, a dilatation of the caudate vein is found, with a diameter of >3mm being indicative of diagnosis in the absence of heart failure.315
1.1.22Intrahepatic veno-venous collateralsThese are usually visualised better with colour/weighted Doppler as small chaotically-distributed vessels around the thrombosed hepatic veins (spider web).307 Les commonly, there are larger vessels that connect two different hepatic veins or a hepatic vein and the inferior vena cava via an uncommon route (e.g., subcapsular or intracapsular).316 The presence of veno-venous collaterals can cause flow inversion in one or more hepatic veins in their permeable sections.307
Portosystemic and portal vein collateralsIn patients with acute BCS, hepatofugal flow is frequently observed in the portal vein307; thus, in patients with sub-acute or chronic BCS it is common to observe repermeation of the paraumbilical vein (due to intrahepatic PHT), and other signs of PHT can develop such as splenomegaly and portosystemic circulation, as in cirrhotic PHT.
Both CT and MRI offer an excellent map of extrahepatic vessels of collateral circulation, which cannot usually be examined in their entirety via ultrasound.317 However, they are less precise than Doppler ultrasound in detecting small intrahepatic and subcapsular vessels of collateral circulation, which are characteristic of this syndrome.310,318
The coexistence of PVT should always be evaluated in patients with BCS, since this is common (15–20% of cases) and is associated with a poorer prognosis.
Nodules in Budd–Chiari syndromeBenign nodules of a regenerative nature, usually multiple nodules, commonly appear in chronic BCS.319–321 Histologically they are similar to focal nodular hyperplasia or focal nodular hyperplasia-like nodules.322 The majority are arterialised nodules that show contrast uptake in the arterial phase using contrast imaging techniques (CT, MRI and contrast ultrasound) and classically have been described as continuing to have hyper- or iso-uptake in the venous phases.14,16,323 However, the appearance is heterogeneous and there may be different uptake patterns in the imaging tests. A characteristic finding is the presence of a central scar, generally when the nodules are larger than 1cm.16,324 The fact that these nodules are hypervascular requires a differential diagnosis from hepatocellular carcinoma. This has especially been described in congenital forms caused by membranes in the inferior vena cava, an uncommon form in Europe.325,326 However, Moucari et al.14 report a prevalence of hepatocellular carcinoma in BCS similar to that which exists in other chronic liver diseases. This is a retrospective study, and there are no prospective studies with long series that allow for these data to be confirmed. MRI with hepatobiliary excretion contrast can be helpful in diagnosis,321,327 but more studies are needed. To date, there is no established guideline for managing nodules associated with BCS; it is important to know that these are lesions that can grow, generally slowly, although in some cases they can grow quickly without being associated with malignancy.
Venography is not usually necessary to establish the diagnosis of BCS, although it could be helpful in determining the extent of the thrombosis and is useful for measuring pressures and planning treatment. In addition to verifying the obstruction of the suprahepatic veins or the inferior vena cava, it offers a characteristic pattern of opacification by filling the small intrahepatic collaterals (spider web) or subcapsular collateral circulation vessels.328
Radiology in idiopathic portal hypertensionIn an ultrasound scan, the liver in IPHT may be indistinguishable from a cirrhotic liver,329 but more commonly the liver surface is only slightly irregular (without clear nodules), and the liver can have an atypical morphology with marked subcapsular segmental hypertrophy (visible as an approach of the hepatic veins and portal branches to the hepatic capsule).330 In patients with IPHT a large splenomegaly should be notable.331 Using portography, abnormalities have been reported in the branches of the portal vein (decreased medium-sized branches, irregularities and interruptions at this level, and subcapsular areas without portal vascularisation),332 and in one study it has been postulated that 3D ultrasound with contrast medium can reproduce this finding, making it possible to differentiate IPHT from cirrhosis.333 In Asian populations, specific changes in IPHT have also been reported after ultrasound contrast medium (Sonazoid), that correspond to a delay in the periportal thickening.334,335 There are no data in European patients, and these techniques must be considered experimental. In MRI, an increase in the intensity of the periportal signal has been described in T2-weighted sequences336–338 and a periportal enhancement in the hepatobiliary phase of the liver-specific contrast Gd-EOB-DTPA in the MRI.339 Other less common findings are peripheral hyper-uptake in acute phases338,340 and a higher incidence of FNH-like benign hypervascular liver nodules.59,341
It has been shown that liver rigidity (which reflects the presence of liver fibrosis) via transition elastography (FibroScan)342 or via Point Shear-Wave Elastography (pSWE)343 is significantly inferior in patients with IPHT than in cirrhotic patients, on par with the clinical manifestations. In the two published studies, liver rigidity in IPHT was an average of 8kPa342 and 1.56m/s343; these values are not strictly normal, but are inconsistent with the typical findings of cirrhotic PHT (liver rigidity of at least 13kPa, and normally >20kPa; pSWE >2.4m/s).292
However, the spleen rigidity (which does seem to reflect the severity of the PHT independent of its cause)344 is similarly elevated (or is even higher in IPHT) in both patient groups.343 Although validation data in extensive series are lacking, and there is a broad overlap of values between IPHT and cirrhosis in the two published studies, the measurement of rigidity in the liver (possibly in combination with spleen rigidity) could be useful for differentiating IPHT and cirrhotic PHT.
Radiology in portal vein thrombosisIn cases of clinical suspicion of PVT, the first diagnostic imaging technique to be used is ultrasound since it has a sensitivity and specificity >90% for identifying thrombosis (similar to CT) and is generally sufficient to establish or rule out thrombosis.288 The diagnostic elements of PVT include at least one of the following signs: (1) presence of an endoluminal thrombus in at least one of the portal system vessels; (2) absence of flow in one or more vessels of the portal system, or (3) presence of cavernomas.87
In greyscale ultrasound, the thrombosis is identified as echogenic material inside the lumen of the vessel; it can partially (mural or partial thrombosis) or fully block the vessel.87 Colour/weighted Doppler and pulsed-wave Doppler can complete the examination by confirming the presence or absence of residual venous flow. The absence of flow should be confirmed using the minimum pulse repetition frequency (PRF) possible to avoid confusion with slow flows (particularly in cirrhosis). Ultrasound is equally sensitive and specific for the diagnosis of splenic vein thrombosis, while it is not sufficiently sensitive to diagnose superior mesenteric vein thrombosis.288,345 Therefore, in the case of suspected mesenteric thrombosis (abdominal pain suggestive of intestinal infarction or signs of severity), CT with contrast medium should be used first.
In patients diagnosed with PVT, the ultrasound should provide information on the presence or absence of cirrhosis, since this can modify the therapeutic approach. Given that in thrombosis that has evolved over a long period of time the liver can undergo significant morphological changes (segmental atrophy and hypertrophy of other segments) similar to those which occur in cirrhosis, differential diagnosis via traditional imaging techniques can be difficult or impossible. This aspect has improved with the introduction of elastographic techniques.346
Regarding dating the thrombosis, it is possible to detect calcifications in the thrombus or in the vessel wall via ultrasound (and CT, but not via MRI), suggesting that the thrombosis has existed for a long period of time.87 In patients who develop cavernomas after a complete PVT, ultrasound identifies small tortuous vessels with hepatopetal flow that replace the vessel affected by the thrombosis.87 The clinician should attempt to evaluate and report on the presence and size of a residual portal vein and a dominant vessel within the cavernoma, since this can affect the therapeutic approach (e.g., TIPS).
In patients who develop PVT in the context of a hepatocellular carcinoma, or in those in whom focal hepatic lesions and PVT are detected at the same time, a differential diagnosis between simple thrombosis and tumour vascular invasion should be considered. The terms “malignant thrombosis” and “neoplastic thrombosis” should be avoided when referring to this condition, since they are incorrect. The signs that suggest vascular invasion are: (1) interruption of the portal vein wall; (2) widening (>2cm) of the vessel, and (3) the presence of arterial hypervascularisation in the event of invasion by a hepatocellular carcinoma or cholangiocarcinoma.137,347,348 Intravascular tumour tissue tends to display behaviour with the contrast which is similar to that of the primary tumour tissue, showing hyper-uptake in the arterial phase and early washout in the late phase. Ultrasound with contrast medium is useful for detecting this behaviour (high specificity)137,289,347,348 and a test with contrast (ultrasound, CT or MRI) should be used in patients with cirrhosis, presence or history of hepatocellular carcinoma and recent-onset PVT.290,349
In addition to the extent of the thrombus, CT and MRI can provide more information than ultrasound about other causes of thrombosis beyond cirrhosis (pancreatitis, abdominal septic foci) and, in cases where the mesenteric veins are affected, they can assess signs of intestinal ischaemia.
In chronic thrombosis with cavernoma formation, MR cholangiography is the technique of choice to evaluate the presence of PC, which is present in more than 80% of patients with cavernomas.97
Performing at least one MR cholangiogram at the time of diagnosis is recommended for all patients with cavernomas. In acute thrombosis in which no recanalisation is achieved, it is also recommended to perform an MR cholangiogram 12 months after the acute episode to check for PC. If no severe cholangiopathy (degree 3) has developed during this period, the probability of progression is low and it does not seem necessary to carry out further evaluation studies.96
Radiology in hereditary haemorrhagic telangiectasiaColour Doppler ultrasound is the first-level technique to assess liver involvement in patients with suspected hereditary haemorrhagic telangiectasia (HHT) and in monitoring of patients with asymptomatic HHT, since a good correlation with the clinical course of the disease has been demonstrated.235 However, the sensitivity of this technique seems to be inferior to that of CT,242 and the role of other techniques is rapidly being redefined.
There are many ultrasound signs of liver involvement in HHT.239,350–352 The most important vessel to evaluate in patients with HHT is the hepatic artery since changes to it reflect the presence of liver involvement at an early stage.239
B-mode should be used to evaluate the diameter and extrahepatic and intrahepatic morphology of this vessel and pulsed-wave Doppler should be used to measure flow velocity and resistive indices. Liver involvement progressively leads to an increase in hepatic artery diameter, greater tortuousness of the vessel in its extrahepatic and intrahepatic parts, and to accelerated flow and low resistance. Also, small parenchymal telangiectasias (colour spots) can be observed via colour/weighted Doppler, in particular with high-frequency probes at the subcapsular level.221 In more advanced phases of liver involvement, dilatation of the hepatic and portal veins can appear (including inversion of portal flow). There have been cases of patients with HHT developing liver cirrhosis. In this phase, the liver can have the classic ultrasound signs of this disease (nodular surface, heterogeneous parenchyma).239,350–352 Finally, focal liver lesions can be found in patients with HHT; most of them are focal nodular hyperplasias and should be more extensively studied using MRI (see below).
To simplify ultrasound diagnosis of liver involvement in patients with HHT, using the combination of criteria in Table 11 has been suggested.225 Two major criteria or one major and two minor criteria would be sufficient to diagnose liver involvement in HHT. Liver changes have been described in 74–100% of CTs of patients with this syndrome, the majority of which are clinically silent.222,228,353 Despite the fact that Doppler ultrasound is the first-line technique, CT is gaining increasing importance in the management of the disease for establishing the diagnosis. The role of CT in managing this disease is not well established, and some authors only recommend it when liver involvement due to vascular malformations is severe with major haemodynamic impact and clinical repercussions, or when associated with focal liver lesions for characterisation. CT, however, allows for detection of signs of the disease that are not possible to detect with ultrasound and that could be useful in diagnosis and even in determining the degree of involvement.240 It also allows for detection of vascular malformations in other abdominal organs, such as the pancreas.241,242 More studies that correlate CT findings with clinical evolution are necessary.
Simplified ultrasound criteria for the diagnosis of liver involvement in hereditary haemorrhagic telangiectasia225
| Criteria | Ultrasound findings |
|---|---|
| Major | Dilatation of the common hepatic artery>7mm |
| Intrahepatic arterial hypervascularisation | |
| Minor | Peak systolic velocity in common hepatic artery>110cm/s |
| Common hepatic arterial resistive index<0.60 | |
| Maximum portal vein flow velocity>25cm/s | |
| Tortuous common hepatic artery | |
| Optional (not used for diagnosis) | Portal vein dilatation>13mm |
| Hepatic vein dilatation>11mm | |
| Hepatomegaly>15cm (midclavicular line) | |
| Nodular liver surface |
To detect all the signs associated with the disease, it is essential to include an arterial phase in the CT or MRI study. Both techniques allow for detection of the characteristic findings–hepatic artery aneurysms, arterioportal fistulas, arteriovenous fistulas, veno-venous shunts and telangiectasias–with great reliability.222,229,242,353 Arterioportal and arteriovenous fistulas are highlighted in the CT and MRI due to early opacification of the portal or suprahepatic veins during the arterial phase, a finding which cannot be detected with Doppler ultrasound.
Despite having insufficient studies comparing CT to ultrasound, the former seems superior in detecting small vascular anomalies that could go unnoticed in ultrasound, especially small peripheral arteriovenous fistulas or small telangiectasias.242 One of the most specific findings in CT (up to 100% specificity reported) is the presence of dilated and tortuous intrahepatic arterial branches.242
A relatively frequent finding in CT is the presence of confluent vascular masses, which correspond to areas with multiple joined telangiectasias, or large short circuits that are directly visible.222,354,355 These are masses with contrast uptake in the arterial phase that continue to have increased uptake in the venous phases.
A higher prevalence of focal nodular hyperplasia has also been reported in HHT (around 3%),220,229,356 although there is one series reported with prevalences of up to 47%.232 MRI can be useful in these cases; however, its role in the diagnosis of the disease has not been studied in depth. Arteriography is no longer used for diagnosis, being reserved for therapeutic procedures.
Radiology in sinusoidal obstruction syndromeIt is impossible to establish a diagnosis of sinusoidal obstruction syndrome (SOS) using imaging techniques, since there are no specific signs. Nevertheless, they provide data that can support the clinical suspicion and rule out other possible diagnoses.
B-mode ultrasound is the first-line test. Hepatomegaly and ascites and thickening of the bile duct wall (>6mm) are usually observed in patients with SOS, and dilatation of the biliary tree can be ruled out.357
The large increase in sinusoidal pressure leads to haemodynamic changes and, via colour and pulsed-wave Doppler, increased resistive indices in the hepatic artery can be observed at an early stage, later observing an inversion in portal flow (initially segmental or up to the main portal) and a decrease in flow in hepatic veins. The combination of these ultrasound criteria had prognostic value in one study.358 The role of hepatic elastography and ultrasound with contrast in SOS is not defined, but an increase in hepatic rigidity has been described due to pSWE and heterogeneous perfusion caused by ultrasound contrast.359
In CT and MRI with conventional contrast the findings are nonspecific: periportal oedema, ascites, decreased diameter of suprahepatic veins (<0.45cm).360–363 A few years ago it was reported that MRI with superparamagnetic iron oxide contrast agents were able to demonstrate a reticular pattern in livers affected by this syndrome, with a sensitivity of 87% and specificity of 89%, even allowing for subclinical detection of the syndrome.275,364 However, these contrast agents are not currently available. In a recent study with liver-specific contrast agents with biliary excretion, gadoxetic acid (Gd-EOB-DTPA, Primovist®), Shin et al.274 report that it is also possible to detect a reticular pattern with high specificity, nearly 100%, although the sensitivity is low and it is not useful for detecting mild forms. This finding has been supported by isolated cases reported,365 without there having been robust studies to date that make it possible to establish the role of MRI with the new liver-specific contrast agents in the diagnosis of this disease.
1.1.23General recommendations- •
Ultrasound (B-mode+colour Doppler and pulsed-wave Doppler) is the initial imaging technique in the study of patients with PHT, in order to establish the permeability of the portal and suprahepatic veins, as well as to evaluate the liver parenchyma (A).
- •
In patients with PHT of an unknown cause and without vascular thrombosis, liver elastography is recommended, since liver rigidity lower than expected in cirrhosis suggests IPHT (C).
- •
To characterise liver nodules in patients with vascular diseases of the liver, MRI is recommended (A).
- •
In patients with vascular liver disease who are potential candidates for a liver transplant, TIPS or surgical portosystemic shunting, performing a CTA and/or MRA to define the vascular anatomy (study of permeable vessels, collateral mapping) is recommended (A).
- •
Semi-annual monitoring with Doppler ultrasound is recommended in all patients with BCS to detect focal lesions, rule out signs of thrombosis progression or confirm the permeability of TIPS (B).
- •
Semi-annual monitoring with Doppler ultrasound is recommended in patients with IPHT to detect asymptomatic PVT (D).
- •
Use of CTA is recommended in the case of suspected intestinal infarction, or when there are no expert abdominal sonographers available (A).
- •
In patients with prior diagnosis or concomitant primitive liver neoplasm, a tumour origin of PVT should be ruled out (B).
- •
T or MRI with contrast are recommended to detail the severity, extent of the thrombosis and to map the collateral circulation (B).
- •
To evaluate the presence and degree of PC, use of MR cholangiography is recommended (A).
- •
In patients with acute PVT who receive anticoagulant therapy, conduct of an imaging test is recommended 6 months after starting treatment to evaluate the degree of recanalisation.
- •
In patients with chronic PVT, conduct of an imaging test is recommended every 6–12 months to evaluate possible rethrombosis phenomena (D).
- •
The chosen imaging technique for monitoring should be individually evaluated based on the location of the thrombosis, the possibility of being visualised by ultrasound and the presence or absence of symptoms (D).
- •
The presence of liver involvement in patients with HHT should be initially evaluated with ultrasound (B-mode+colour Doppler and pulsed-wave Doppler) (A).
- •
In patients with liver involvement due to HHT, the conduct of a CT and/or an MRI with contrast will be considered to obtain a more detailed study of the liver involvement (C).
- •
Annual monitoring of asymptomatic patients with imaging techniques via Doppler ultrasound is recommended (B).
Thrombophilia is understood to be the increase in susceptibility to suffering thrombotic episodes, especially venous ones.366 Thrombophilia may be congenital or acquired and is identified in around 50% of patients with idiopathic venous thromboembolism (Table 12).366–368 The affected individuals may develop an episode spontaneously, but on occasions, additional risk factors concur which act as triggers, such as surgeries, pregnancy, hormone therapy or cancer.366
Prothrombotic states to consider in the aetiological study of splanchnic vein thrombosis.
| Hereditary prothrombotic states |
| Antithrombin deficiency |
| C-protein deficiency |
| S-protein deficiency |
| Factor V Leiden mutation |
| G20210A mutation of the prothrombin gene |
| Acquired prothrombotic states |
| Myeloproliferative neoplasms (polycythaemia vera, essential thrombocythaemia, myelofibrosis) |
| Paroxysmal nocturnal haemoglobinuria |
| Antiphospholipid syndrome |
| Cancer |
| Pregnancy |
| Use of hormonal contraception |
| Inflammatory bowel disease |
| Behçet's disease |
| Other systemic inflammatory diseases |
Hereditary thrombophilia includes deficiencies in coagulation inhibitors (antithrombin, protein C and protein S), increase in coagulation factors and certain genetic mutations such as Factor V Leiden and the 20210 mutation of the prothrombin gene.368 There are other abnormalities, such as congenital dysfibrinogenaemia and various mutations, or increases in coagulation factors, which may also be implicated. The ABO blood group significantly determines the risk of thrombosis, with a 2–4 times increase in relative risk.368
In general, the prevalence of thrombophilic defects in splanchnic thrombosis is higher than that found in other territories in which vein thrombosis are much more frequent and other risk factors are more common. Occasionally, coagulation inhibitor levels are not easy to interpret, given that hepatic synthesis may be decreased and, also, synthesis of protein C and S is vitamin K-dependent. The antithrombin deficiency varies between 0 and 5%, depending on the series; the protein C deficiency, between 4 and 20%, and the protein S deficiency, between 0 and 30%.8,75,117,369–371
The presence of Factor V Leiden mutation is more frequent in the general population. In these patients, a prevalence between 7 and 32% has been reported. In general, they are heterozygous carriers, although they can be homozygous.372
Another frequent mutation involved in thrombophilia is the G20210A mutation of the prothrombin gene (PT20210A). It has been calculated that carriers of Factor V Leiden show a 2–11 times increase in risk and carriers of the PT20120A mutation, between 2 and 5 times.373
Other genetic abnormalities have not been evaluated in these series of patients, but they can also be involved. There is also no information about the role of the ABO group.
1.1.30Acquired thrombophiliaThere are various acquired situations that increase the risk of thrombosis. The most prominent is antiphospholipid syndrome,366–368 caused by antiphospholipid antibodies against cell membranes. Those most related with thrombosis are anticardiolipin antibodies, anti-β2-glycoprotein-I and the lupus anticoagulant. Heparin-induced thrombocytopenia is a potent prothrombotic situation caused by anti-PF4/heparin complex antibodies in patients treated with heparin. There is also an increase in thrombotic risk in sickle-cell anaemia.
In splanchnic area thrombosis, the prevalence of antiphospholipid antibodies has been estimated between 5 and 15%,8,75,117 although, on some occasions, the measurement has not been done following the guidelines to establish that it is an antiphospholipid syndrome, with at least two measurements 12 weeks apart.374
The increase in factor VIII has been related with thrombophilia. The reasons for the increase in factor VIII are not known. There are acute situations that make factor VIII increase, but there are also individuals in which the increase is persistent. There is probably a genetic basis and acquired factors that concur. Higher levels of factor VIII have been found in patients with PVT.375,376 Recently, research laboratories are incorporating global homoeostasis tests, such as thrombin generation, or euglobulin lysis time, although their role is still not currently well established.377
Myeloproliferative neoplasmsMyeloproliferative neoplasms are commonly implicated in the cause of splanchnic thrombosis, especially polycythaemia vera and essential thrombocythaemia. They are clonal diseases of medullary stem cells that are generally characterised by the overproduction of functional mature elements of the three series. One of the main and most frequent complications is the development of arterial or venous thrombosis, caused by platelet hyperaggregability and increased thrombin generation.377 Thrombosis may occur in any area, although they are especially frequent in the splanchnic area, where they are associated with myeloproliferative neoplasms in 30–40% of cases.8,75,370,378,379 On occasion, splanchnic thrombosis occurs in patients who have not yet been diagnosed with myeloproliferative neoplasms, since abnormalities have not yet presented in the peripheral blood counts.
The diagnosis of myeloproliferative neoplasms is based on established criteria that include changes in peripheral blood counts and medullary findings. In patients with splanchnic thrombosis who have hypersplenism due to PHT secondary to the thrombosis, these criteria can be difficult to apply. Especially in these cases it is very helpful to determine specific molecular markers, such as the JAK2V617F mutation, which can be detected in patients with splanchnic thrombosis. In a recent meta-analysis, the prevalence of myeloproliferative neoplasms and the JAK2V617F mutation was 40%, approximately, in BCS and 30% in PVT (Table 2).380 The JAK2V617F mutation in patients with splanchnic thrombosis without peripheral blood abnormalities was found in 17.1% of patients with BCS and in 15.4% of patients with PVT.380 This makes it recommended to rule out the presence of myeloproliferative neoplasms and to determine the JAK2V617F in patients with splanchnic thrombosis. It should be kept in mind that in patients with negative mutations it is sometimes appropriate to repeat the determination, given that it may become positive during evolution due to the increase in the clonal population in peripheral blood. These may also be negative in neutropaenic patients, given that the representative DNA is in the granulocytes and not the lymphocytes. The JAK2V617F mutation is present in practically all patients with polycythaemia vera and in approximately 50% of patients with essential thrombocythaemia or myelofibrosis. There are other mutations involved in myeloproliferative neoplasms that may also be evaluated when the JAK2V617F mutation is negative.26,27 Calreticulin (CALR) mutations are more common (80%) in patients with essential thrombocythaemia and myelofibrosis without the JAK2V617F mutation, and are less common in patients with polycythaemia vera.28
The underlying physiopathological mechanism by which these patients develop a thrombotic episode is not completely known. In addition to thrombocytosis and erythrocytosis, abnormalities in leukocytes and platelets have been identified, which can cause hypercoagulability.381
Paroxysmal nocturnal haemoglobinuriaParoxysmal nocturnal haemoglobinuria is a very rare clonal disease. Patients have a notably increased risk of thrombosis. It is associated more with BCS (9–13%) than with PVT (0–3%).75,382 The underlying mechanism is unknown, but when the clonal population increases above 60%, the risk of thrombosis is very high.383 Paroxysmal nocturnal haemoglobinuria should be ruled out in patients with BCS and be considered in those with PVT.
MiscellaneousThere are other conditions that can be associated with splanchnic thrombosis, although less commonly than those previously mentioned, such as autoimmune diseases, inflammatory bowel disease, vasculitis, sarcoidosis, Behçet's disease, or coeliac disease. Nephrotic syndrome can cause acquired antithrombin deficiency, but the most common thrombosis is in renal veins, especially in childhood.
The use of hormonal contraceptives and pregnancy have been implicated as splanchnic thrombosis risk factors. Particularly, there is a twofold increase in the risk of BCS.30
Relevance of aetiological factors in treatmentThe presence of aetiological factors is very relevant, given that it has therapeutic and prognostic implications. Therefore, it is necessary to investigate them systematically.
In patients with BCS, indefinite anticoagulant therapy is usually recommended due to the severity of the symptoms. In patients with PVT, treatment is usually recommended for 6 months, although in some situations the risk of recurrence is so high that it may also be advisable to maintain it indefinitely, especially in patients with low risk of bleeding.
There are few studies evaluating the risk of PVT recurrence. In these patients the presence of underlying thrombophilia is a risk factor for recurrence.108,109,384 It is also necessary to consider the risk of bleeding, especially from oesophagastric varices. Therefore, recent guidelines have suggested extending anticoagulant therapy in patients with greater thrombophilic factors, such as antithrombin deficiency and those who are homozygous for Factor V Leiden and PT20210A mutations, although there is no evidence of sufficient quality and the recommendation is based on extrapolations from thrombosis in other areas.385,386
In the case of myeloproliferative neoplasms, anticoagulant therapy should be administered indefinitely. Most patients with myeloproliferative neoplasms receive aspirin as primary prevention. It is unknown whether, in the case of requiring anticoagulant therapy, antiaggregant treatment should be continued, although a potential benefit has been identified in retrospective studies.386,387
In addition to antithrombotic treatment, patients with myeloproliferative neoplasms should receive the indicated treatment for their underlying disease.
In patients with paroxysmal nocturnal haemoglobinuria, the presence of a splanchnic thrombotic complication may indicate indefinite eculizumab treatment.388
Recommendations- •
In patients with splanchnic vein thrombosis the existence of systemic (hereditary and acquired thrombophilic factors, myeloproliferative neoplasms, paroxysmal nocturnal haemoglobinuria, autoimmune diseases) and local (neoplasms, inflammatory processes, recent surgeries) prothrombotic factors should be investigated.
- •
The evaluation of thrombophilia should include determination of antithrombin, protein C, protein S, Factor V Leiden mutation, PT20210A mutation, lupus anticoagulant and antiphospholipid antibodies. If these last two are positive, they should be re-evaluated at 12 weeks.
- •
Determining the JAK2V617F and calreticulin mutations is recommended to rule out myeloproliferative neoplasms, including in patients with normal peripheral blood counts. If the mutations are negative, considering a medullary cytohistological study is recommended. In case of suspicion of myeloproliferative syndrome, evaluation and monitoring by a haematologist is recommended.
- •
Frequently various factors coexist, thus the finding of one factor does not rule out the existence of others.
After diagnosis of BCS, patients should receive anticoagulant therapy as soon as possible in order to reduce the extent of the thrombus and prevent new thrombosis.1,8,15,389 If repermeation is not achieved with anticoagulant therapy or the symptoms so require it, recanalisation of the flow in suprahepatic veins should be considered using angioplasty, placement of endovascular prostheses, thrombolysis or TIPS.
Given the lack of specific evidence in this area, the recommendations established for deep vein thrombosis or pulmonary embolism are used.390 They should receive anticoagulant therapy with low-molecular-weight heparins (LMWH) at therapeutic doses for at least 5–7 days followed by vitamin K antagonists with the doses necessary to maintain INR between 2 and 3. They should overlap for 3–5 days and suspend LMWH when the INR is in therapeutic range for 2 consecutive days. There is no reported experience about the use of direct-acting oral anticoagulants in this condition.
The risk of haemorrhage in patients with BCS is high (17% in recent series). It is therefore very important to precisely manage the anticoagulant therapy and prevent PHT-related bleeding.39
Maintaining anticoagulant therapy at an early stage is recommended in patients with BCS who required a liver transplant in order to prevent recurrences. The only exception could be in thrombophilic states, given that they are corrected by liver transplant.
Acute portal thrombosisThe main objectives in treatment of acute PVT are: (1) preventing spreading of the thrombosis to the mesenteric veins and, therefore, mesenteric venous infarction, and (2) recanalisation of the portal vein.
AnticoagulationIn a recent wide cohort, early starting of anticoagulant therapy prevented spreading of the thrombus.75 Limited mesenteric infarction was confirmed in 2/95 patients, although 60% of the patients had initial involvement of the superior mesenteric vein. Recanalisation was achieved in the portal vein in 39% of cases, in the splenic vein in 80% and in the mesenteric vein in 73% of patients on anticoagulants for more than 6 months. Haemorrhage occurred during the period of anticoagulant therapy in 9% of patients. Factors related to recanalisation failure were splenic vein obstruction, ascites and delayed start of anticoagulant therapy.
The most used first-line drugs were heparin and LMWHs at therapeutic doses, and later vitamin K antagonists with an INR between 2 and 3. LMWH is preferred over unfractionated heparin due to the lower incidence of heparin-induced thrombocytopenia that has been identified more frequently than expected in patients with acute PVT.391 There is no experience regarding the efficacy and safety of direct-acting oral anticoagulants for this indication.
Thrombolysis does not achieve higher recanalisation rates than anticoagulant therapy and involves a higher risk of bleeding. Therefore, it is not recommended.
Currently, recanalisation techniques via angioplasty or endovascular prosthesis are being used which offer good results, especially in post-surgical thrombosis.107
Other causes of extrahepatic portal vein obstructionExtrahepatic portal vein obstruction is due to three mechanisms: neoplastic invasion, compression by tumour mass, or portal vein cavernoma.
In this situation, the evidence of the benefit of anticoagulant therapy is poor, with no existing prospective studies on this topic. There are retrospective cohort studies in patients with non-cirrhotic PVT in which the use of anticoagulants has been associated with a reduction in the risk of rethrombosis, with a reduction between 40% and 80%.108,109 The risk of haemorrhage was not increased if the prophylaxis for bleeding was adequate. In one study, anticoagulant therapy was related to a favourable impact on survival with a significant decrease in mortality,392 although this benefit was not observed in another more recent study.109
Management of anticoagulant therapy in patients with liver diseaseThe most widely used guidelines on antithrombotic treatment are ‘The American College of Chest Physicians Evidence-Based Clinical Practice Guidelines’ (9th Edition).390 There is a recent update which includes existing evidence with direct-acting oral anticoagulants, but it is only considered for deep vein thrombosis of the lower limbs and pulmonary embolism.393 In splanchnic thrombosis, the current recommendations continue to be those from the 9th edition.390 These guidelines, for splanchnic thrombosis, only include LMWHs and fondaparinux as first-line treatment. These are anticoagulants that intensify the anticoagulant effect of antithrombin by inhibiting factor Xa and thrombin (fondaparinux only inhibits Xa). As long-term treatment, it considers vitamin K antagonists.
HeparinsUnfractionated heparin is effective as first-line treatment, but it has a higher risk of thrombocytopenia and osteoporosis; it must be monitored via activated partial thromboplastin time (APTT) and the therapeutic ranges are not well standardised. Due to these limitations, use of LMWHs is preferable; they can be administered subcutaneously 1 or 2 times per day and do not require routine monitoring, except in some cases (pregnancy, kidney failure and obese patients).394
The biggest problem in these patients is that the effect of these drugs depends on antithrombin, which is synthesised by the liver and is decreased in patients with advanced disease. This results in the question arising of whether monitoring is necessary in these patients to adjust the dose, although there is insufficient evidence to recommend adjusting the dose based on anti-Xa levels.395,396
Vitamin K antagonistsVitamin K antagonists (VKAs) are coumarin drugs that interfere in the carboxylation of vitamin K-dependent factors (factor VII, IX, X and prothrombin) and of anticoagulation proteins C and S. Their therapeutic range is small and they require periodic monitoring to adjust the dose.397 There are also important inter-individual variations due to genetic polymorphisms and drug and dietary interactions that complicate its management. The test of choice to monitor its effect is prothrombin time, expressed as INR. INR includes an adjustment which makes it independent of the measurement instrument or reagent used.398 This adjustment determines that the prothrombin time values expressed as INR are only applicable to patients receiving VKAs and not to patients with liver disease or lacking factors for a variety of reasons (consumption, loss through haemorrhage, hereditary). The therapeutic range in patients with venous thromboembolism (including splanchnic vein thrombosis) is an INR of 2–3.
The main concern with use of VKAs in liver disease is the possibility that the baseline prothrombin time will be prolonged. This would mean a possible underdosage, although there are no studies on this topic. The baseline prothrombin time may also vary depending on the liver disease evolution and increase the instability of the controls. In patients with liver disease, use of VKAs is complex and their efficacy and safety may be compromised.
Direct oral anticoagulantsThese are oral anticoagulants that act by specifically inhibiting coagulation factors. Dabigatran inhibits thrombin and rivaroxaban; apixaban and edoxaban inhibit activated factor X. They have a short half-life and a rapid onset of action, their pharmacokinetics are foreseeable, they do not require routine monitoring, have few drug interactions and are not affected by ingestion. Therefore, they have advantages over VKAs. Recently, they have been approved as first-line and long-term treatment of venous thromboembolism with an efficacy profile similar to that of conventional treatment, although with a better safety profile.393 Although they may have theoretical advantages in patients with liver disease, cirrhosis drugs have been excluded from the main clinical trials, and therefore should not be used at the moment. In splanchnic thrombosis, there are some cases treated on an exceptional basis, but they should not be used until there is more specific information available in these patients.
Recommendations- •
In cases in which anticoagulant therapy is indicated, starting treatment with LMWH is recommended.
- •
LMWH is used in fixed doses as prophylaxis or weight-adjusted for therapeutic doses.
- •
Patients receiving treatment with LMWHs should be monitored for the onset of thrombocytopenia. It is not necessary to monitor anti-Xa levels, except in pregnant women, obese patients or those with kidney failure.
- •
The temporary suspension of anticoagulants should be considered before any invasive procedures, including paracentesis.
- •
The duration of anticoagulant therapy should be at least 6 months.
- •
Once the gastrointestinal bleeding prophylaxis is administered, it is recommended to consider permanent anticoagulant therapy in patients with relevant prothrombotic situations, history of intestinal ischaemia or recurrent thrombosis.
- •
In patients with underlying myeloproliferative neoplasms, long-term anticoagulant therapy is indicated.
- •
If permanent anticoagulation is considered, VKA treatment is recommended. It must be administered at the doses necessary to maintain INR levels between 2 and 3, although in cirrhosis they may not be representative.
- •
Currently, there is insufficient evidence about the efficacy and safety of direct-acting oral anticoagulants to recommend their use in patients with cirrhosis or splanchnic thrombosis.
The authors declare that they have no conflicts of interest.
Please cite this article as: Martín-Llahí M, Albillos A, Bañares R, Berzigotti A, García-Criado MÁ, Genescà J, et al. Enfermedades vasculares del hígado. Guías Clínicas de la Sociedad Catalana de Digestología y de la Asociación Española para el Estudio del Hígado. Gastroenterol Hepatol. 2017;40:538–580.
