


La queratosis pilarse agrava con el usode jabón, de agua caliente,contacto con ropa de lanay durante el invierno
El estrato córneo es la capa más superficial de la piel y su formación es el resultado final de un proceso metabólico llamado queratinización. Cuando nos enfrentamos a un paciente con desórdenes en el número de células de la piel que se multiplican, o con una alteración en la diferenciación celular y/o en su posterior eliminación normal en la superficie cutánea, hablamos de alteraciones de la queratinización, que pueden ser tanto hereditarias como adquiridas, generalmente secundarias en este caso a otras enfermedades. Se manifiestan clínicamente en forma de escamas o masas queratósicas localizadas o generalizadas en la superficie cutánea. Constituyen un grupo amplio dentro del cual cabe hacer referencia a las ictiosis, la queratosis pilar y las queratodermias palmoplantares como principales trastornos de la queratinización.
DERMATOSIS ICTIOSIFORMES
Ictiosis es el nombre que se aplica a un grupo de alteraciones caracterizadas por una descamación no inflamatoria generalizada y persistente de la superficie cutánea. Tienen en común la aparición de piel seca, cuarteada y recubierta de escamas que recuerdan a los peces (ictiosis deriva de la palabra griega que significa pez). La gran mayoría son congénitas (determinadas genéticamente) pero también hay formas adquiridas.
Se caracterizan por su curso crónico, por empeorar en invierno y en climas secos y presentar una expresión clínica variable de un sujeto a otro.
ICTIOSIS CONGÉNITAS
Entre las ictiosis congénitas cabe distinguir la vulgar, la ligada al cromosoma X, la laminar y la eritrodermia ictiosiforme ampollosa. En el recién nacido se habla de bebé colodión y feto en arlequín. Se decriben estas formas a continuación, junto a otras menos comunes.
Ictiosis vulgar
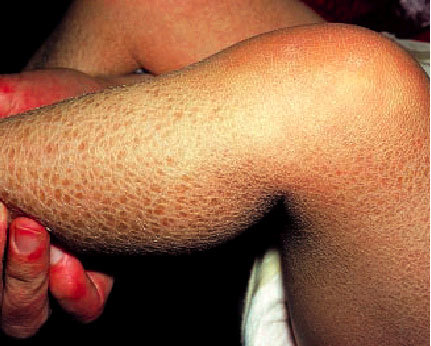
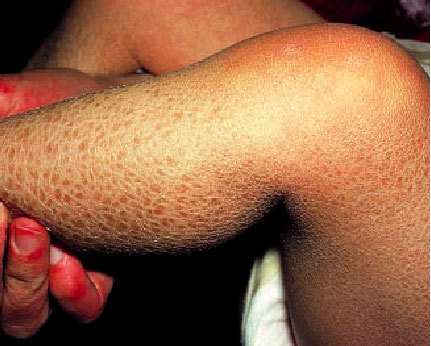
Representa el trastorno de la queratinización más frecuente (imagen de apetura). Tiene una incidencia de 1 por cada 300 habitantes y su modalidad de transmisión es de forma autosómica dominante y afecta por igual a ambos sexos. Su patogenia está relacionada con un trastorno de la síntesis de profilagrina (y queda como resultado una menor hidratación del estrato córneo) que se asocia con un tiempo de tránsito celular epidérmico normal.
Suele comenzar en una fase temprana de la niñez y, en general, no se encuentra presente al nacer. Se caracteriza por la presencia de escamas blanquecinas, finas, de pequeño tamaño y de bordes generalmente invertidos que cubren la superficie cutánea de forma generalizada pero más acentuada en las superficies extensoras de las extremidades respetando las zonas de flexión. Es frecuente la presencia de una acentuación de los pliegues cutáneos de palmas y plantas, así como una queratosis pilar de los brazos, los muslos y las nalgas. A menudo, se ha asociado en estos pacientes la presencia de eccema atópico, asma o urticaria. La ictiosis vulgar es la forma más benigna, presenta exacerbaciones con el clima frío y seco y mejora durante el verano. Este trastorno dura toda la vida y en general mejora de forma progresiva en la vida adulta
Para el diagnóstico son importantes (tanto en ésta como en otras ictiosis) los antecedentes familiares, personales y el examen físico. El estudio histopatológico de la biopsia de piel en estos pacientes revela una hiperqueratosis con ausencia de la capa granular de la epidermis. El tratamiento es similar en todas las ictiosis y se comentará más adelante.
Ictiosis ligada al cromosoma X
Es una enfermedad poco frecuente: su incidencia es de 1 por cada 6.000 hombres y es de herencia recesiva ligada al cromosoma X. Se debe al déficit del gen de la enzima sulfatasa de esteroide localizado en el extremo distal del brazo corto del cromosoma X. Afecta fundamentalmente a los varones, las mujeres son portadoras y pueden mostrar alguna manifestación aislada.
La ictiosis aparece generalmente entre la primera y tercera semana de vida, y se encuentra presente en todos los casos antes del año de edad. Las escamas tienden a ser grandes, gruesas, adherentes y de color oscuro. Afecta preferentemente las superficies extensoras, los pliegues, las orejas, el cuello y el cuero cabelludo. Respeta las zonas palmoplantares y no se observa queratosis pilar ni atopia. Debido a la coloración marronácea de las escamas, estos pacientes tienen el aspecto de estar sucios. Se observan además opacidades corneales y alteraciones en los lípidos plasmáticos. Dura toda la vida y no mejora con la edad, se agrava en los climas fríos y secos y se observa mejoría en los climas húmedos y calientes.
En estos pacientes se puede realizar un estudio bioquímico determinando la actividad de la sulfatasa de esteroide en fibroblastos, leucocitos, piel total o escamas. Se puede efectuar un diagnóstico prenatal determinando los niveles de sulfatasa de esteroide en células del líquido amniótico. En la biopsia cutánea se observa una hiperqueratosis compacta con presencia de la capa granular.
Ictiosis laminar
Esta forma de ictiosis es poco frecuente: su incidencia es de 1 por cada 300.000 habitantes. Se hereda de forma autosómica recesiva, aunque se ha descrito ocasionalmente de forma autosómica dominante en algunas familias. Comienza en el momento del nacimiento y suele presentarse en forma de «bebé colodión» (véase más adelante). Los niños nacen rodeados de una membrana transparente, lisa y brillante que les confiere un aspecto de barnizado. Si esta fase es superada, se produce una descamación generalizada con escamas compactas, de gran tamaño y con una consistencia de «pergamino». Afecta la totalidad del cuerpo con predominio en las áreas de flexión (fig. 1). Las escamas presentan bordes sobreelevados y adherencia central, se desprenden en abundancia a diferencia de la ictiosis vulgar y la ligada al cromosoma X. Las palmas y las plantas presentan engrosamiento de la capa córnea (hiperqueratosis) y acentuación de los pliegues cutáneos. Alrededor de las grandes articulaciones la piel tiende a ser verrucosa.
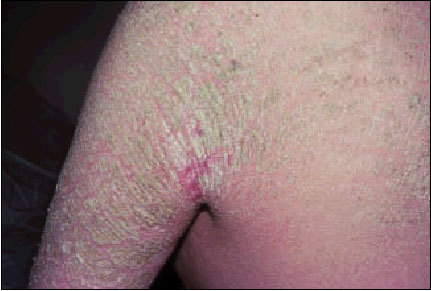
Fig. 1. Ictiosis laminar
El uso de retinoidesorales se restringe a los casos de ictiosismás graves
Es característica la presencia de eversión de la conjuntiva palpebral (ectropion), de eversión de los labios (eclabium) y de pabellones auriculares dismórficos. Pueden presentar alteraciones del pelo y las uñas, fisuras cutáneas e infecciones bacterianas. Debido a la obstrucción de las glándulas sudoríparas estos pacientes no sudan, lo que les puede causar hipertermia en climas calurosos y durante el ejercicio. Esta enfermedad persiste durante toda la vida del paciente.
Eritrodermia ictiosiforme ampollosa
Su incidencia es de 1 por cada 300.000 habitantes. Su herencia es autosómica dominante. Se debe a una alteración en los genes que codifican diversas queratinas con una consecuente vacuolización de la capa granulosa. Se presenta al nacer o poco después del parto. Puede ser generalizada, localizada o palmoplantar. Se observa eritema y ampollas de gran tamaño localizadas frecuentemente en áreas sometidas a traumatismos, que evolucionan hacia una denudación de la piel con el riesgo de infección y sepsis secundaria. Con el transcurso del tiempo, la piel se torna queratósica y verrucosa sobre todo en áreas de flexión, rodillas y codos. Las lesiones presentan un color oscuro y el paciente a menudo despide un olor desagradable debido a sobreinfecciones bacterianas. Las ampollas pueden recurrir en forma periódica durante la edad adulta en un 20% de los pacientes afectados. Se puede realizar diagnóstico prenatal con fetoscopia y biopsia cutánea. Es la ictiosis más refractaria al tratamiento.
Formas del recién nacido
Entre ellas cabe citar el bebé colodión y el feto en arlequín.
Bebé colodión
Es la expresión clínica neonatal de diferentes ictiosis (ictiosis laminar, eritrodermia ictiosiforme ampollar, ictiosis ligada al cromosoma X y excepcionalmente ictiosis vulgar). El bebé colodión presenta incapacidad de succionar, dificultad en regular su temperatura y dificultad respiratoria debido al efecto restrictivo de la gruesa membrana que lo envuelve. El aspecto final depende del origen de la afectación.
Feto en arlequín
Es la forma congénita más severa que existe, denominándose también ictiosis congénita maligna. El niño presenta toda la piel cubierta por grandes placas córneas muy gruesas y separadas por fisuras profundas, que originan un patrón en forma de rombos en la superficie de la piel lo que se ha asemejado con un traje de arlequín. Los rasgos faciales están muy deformados. Generalmente se produce la muerte en las primeras semanas de vida.
Formas menos comunes
Existen otras ictiosis congénitas de muy baja frecuencia que presentan manifestaciones sistémicas asociadas a la afectación cutánea:
Enfermedad de Refsum: presenta una ictiosis similar a la vulgar, ataxia cerebelosa, retinitis pigmentosa y neuropatía periférica.
Síndrome de Netherton: ictiosis lineal circunfleja asociada a un defecto en el tallo del pelo.
Síndrome de Rud: ictiosis lamelar, enanismo, retraso mental o hipogonadismo.
Síndrome de Sjögren-Larsson: ictiosis lamelar discreta, parálisis espástica y degeneración macular de la retina.
Enfermedad de Conradi: descamación en espiral, atrofia folicular, alteraciones esqueléticas y opacidades corneales.
ICTIOSIS ADQUIRIDAS
La ictiosis adquirida no es frecuente, es similar a la vulgar, y se observa en pacientes con enfermedades malignas tales como enfermedad de Hodgkin, no-Hodgkin, micosis fungoide, mieloma múltiple, cáncer de mama, pulmón, leiomiosarcoma, sarcoma de Kaposi. También se puede presentar asociada a enfermedades no malignas como trastornos metabólicos crónicos (malabsorción, malnutrición), falla renal, conectivopatías o enfermedades granulomatosas.
EVOLUCIÓN Y PRONÓSTICO
Las dermatosis ictiosiformes persisten durante toda la vida, rara vez dan lugar a problemas médicos graves, aunque sí pueden aparecer trastornos psiquiátricos como consecuencia de lesiones deformantes.
Es posible efectuar un diagnóstico prenatal en la ictiosis ligada al cromosoma X y en la eritrodermia ictiosiforme ampollar. Es importante ofrecer asesoramiento genético a los niños afectados, a sus progenitores y a mujeres portadoras de ictiosis ligada al cromosoma X.
XEROSIS
Se utiliza el término xerosis o xerodermia para referirse a la sequedad de piel, que es especialmente frecuente en personas mayores y en atópicos. Se caracteriza por una descamación persistente y generalizada que se puede acompañar de prurito. Generalmente empeora en invierno con las bajas temperaturas y en ambientes con baja humedad, así como con los baños repetidos y el secado vigoroso con la toalla. Aunque no se comentará in extenso, es un motivo frecuente de consulta y en su tratamiento es fundamental la hidratación de la piel.
TRATAMIENTO
Es similar en todas las ictiosis, el paciente debe comprender que el manejo es sintomático y que debido a la evolución crónica de estas enfermedades se debe mantener un tratamiento continuo. Procurar ambientes húmedos y cálidos, utilizar jabones suaves y evitar las duchas repetidas que resecan la piel y el roce con prendas de vestir ásperas.
Hidratación
Es un pilar básico en el manejo de estos pacientes. Se debe evitar la evaporación del agua de la capa córnea y alcanzar el máximo efecto hidratante. Con este objetivo se utilizan emolientes de forma continuada, preferiblemente en una base de ungüento, como la vaselina.
Queratolíticos
Facilitan la eliminación de las escamas a través de la separación de las células del estrato córneo, su uso es adicional a los emolientes. Se utiliza ácido láctico en concentraciones del 3 al 12%, lactato amónico o ácido glicólico. Se observa mayor eficacia cuando son aplicados después de hidratar la piel. También se utiliza propilenglicol al 60%, urea al 10-20% y ácido salicílico a bajas concentraciones (2-6%), según el grado de engrosamiento de la piel y acumulación de escamas de la zona a tratar, en una base de ungüento.
Retinoides orales
Su uso se restringe a los casos más graves. Se utiliza la isotretinoína, el etretinato o la acitretina en dosis más altas que las utilizadas en otras patologías. Requieren un estrecho control médico debido a sus efectos secundarios.
Manejo de sobreinfecciones
En el manejo de las sobreinfecciones se utilizan antisépticos tópicos, antibióticos orales y antimicóticos, según necesidad.
El bebé colodión y el feto en arlequín requieren una vigilancia estrecha en una unidad de cuidados intensivos neonatales.
QUERATOSIS PILAR
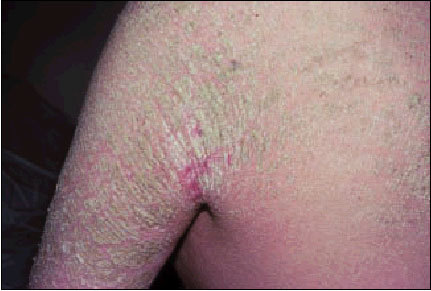
Es un trastorno de la queratinización, frecuente, de causa desconocida. Se asocia con la ictiosis vulgar y es común en pacientes con piel seca. Se presenta el folículo piloso dilatado con un tapón de queratina en su desembocadura. Comienza en una etapa temprana de la infancia o la adolescencia y se caracteriza por pápulas pequeñas como «cabezas de alfiler» en las zonas laterales de los brazos, los muslos y los glúteos. A menudo están rodeadas de un área eritematosa que confiere a la piel un aspecto granulado y un tacto rasposo. Se agrava con el uso de jabón, de agua caliente, contacto con ropa de lana y durante el invierno. Mejora durante el verano. Las lesiones usualmente disminuyen con la edad. El manejo tópico con emolientes generalmente es suficiente. En casos refractarios se utilizan queratolíticos.
QUERATODERMIAS PALMOPLANTARES
Constituyen un grupo heterogéneo de afecciones que tienen en común el producir un engrosamiento de la capa córnea de palmas y plantas de forma localizada o difusa. Afectan a ambos sexos y tienen una distribución mundial, sin diferencia racial; su incidencia es de 1 por 40.000 habitantes. Las queratodermias palmoplantares (QPP) hereditarias son procesos primarios con hallazgos clínicos y genéticos específicos aislados o también asociados a otros trastornos hereditarios de la queratinización. Se han descrito diversos síndromes de QPP asociada a sordera neurosensorial, predisposición familiar a diversas neoplasias y defectos de dientes y uñas. Existen QPP hereditarias con patrón dominante y recesivo, entre las cuales cabe destacar las siguientes:
Mal de Meleda
Es una enfermedad autosómica recesiva severa. Se ha descrito en la isla de Meleda, en el mar Adriático, donde es relativamente frecuente debido a la endogamia de sus habitantes.
Suele presentarse durante las primeras semanas o meses de vida, inicialmente como un eritema palmoplantar, seguido de una gran masa queratósica difusa amarillo parduzca que incluso pueden provocar contracturas. Presentan un aumento patológico de la sudación (hiperhidrosis), con un olor fétido de forma frecuente, alteraciones de las uñas y acortamiento anormal de los dedos de manos y pies.
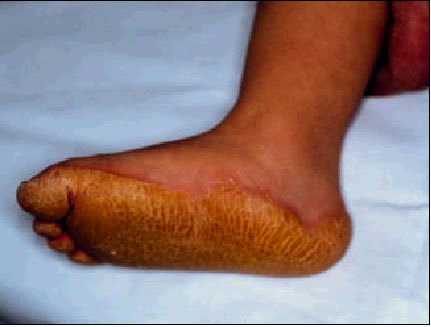
Queratodermia de Unna-Thost
Es de herencia autosómica dominante, se caracteriza por un engrosamiento difuso, simétrico y bien delimitado de las palmas y plantas. Aparece durante los 2 primeros años y persiste durante toda la vida del paciente. Suele asociar hiperhidrosis importante que favorece la presencia de infecciones micóticas. No se observan alteraciones de pelo, uñas ni dientes (fig. 2).
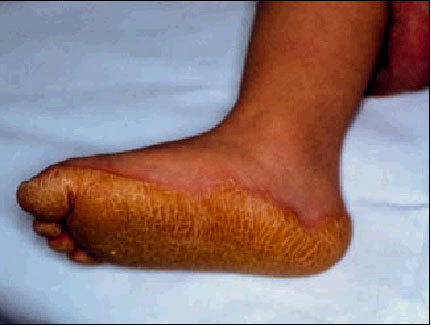
Fig. 2. Queratodermia plantar
La hidrataciónes un pilarbásico en el manejode los pacientes con ictiosis
Queratodermia mutilante
Enfermedad de herencia autosómica dominante. Se inicia en la infancia y se caracteriza por una queratodermia difusa y de superficie irregular que recuerda un panal de abejas. En la edad adulta aparecen bandas fibrosas en manos y pies que provocan compresión de los tejidos, constricción, lo que lleva a una progresiva estrangulación y amputación espontánea de las falanges con el transcurso del tiempo. Se asocia con sordera, alopecia y retardo mental.
Síndrome de Papillon-Lefevre
Enfermedad de herencia autosómica recesiva. Se caracteriza por la asociación de anormalidades dentarias (gingivitis y pérdidas de piezas dentales) con queratodermia palmoplantar difusa (eritrodermia, hiperhidrosis y mal olor).
TRATAMIENTO
El manejo de estas enfermedades es similar al de los otros trastornos de la queratinización. En estos pacientes, dado el grosor de la capa córnea de la superficie palmar y plantar, se requiere un refuerzo en la concentración de los queratolíticos, los que habitualmente sólo mejoran parcialmente la hiperqueratosis. El tratamiento quirúrgico es útil en queratodermias mutilantes. *